
Frequently Asked Questions
Leukemia
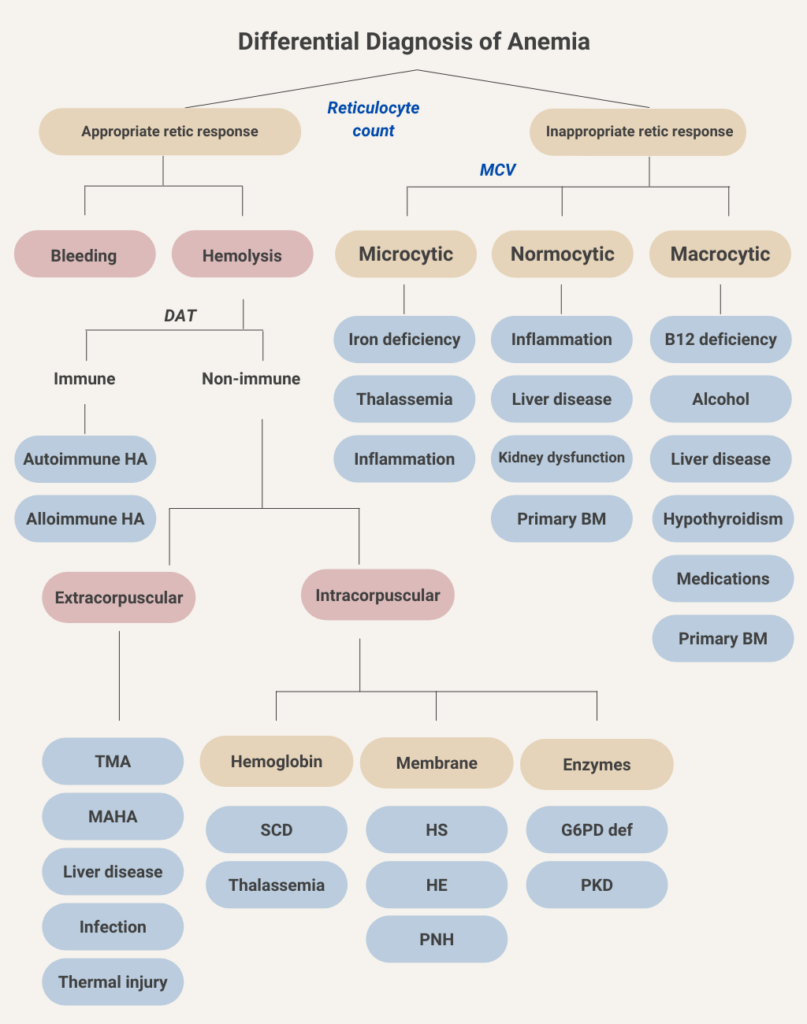
Absolute monoclonal B cells count ≥ 5 × 109 /L (≥ 5,000 cells/mm3) + CLL Immunophenotype in peripheral blood.
Unexplained fever > 38 degrees C (100.4 degrees F) for ≥ 3 days, weight loss of over 10% body weight in prior 6 months, and drenching night sweats.
Chronic lymphocytic leukemia
Hairy cell leukemia
Hematopoietic stem cell transplantation
Monoclonal B-cell lymphocytosis
Myeloproliferative neoplasm
Smudge cells are remnants of fragile lymphocytes that are generated during preparation of peripheral smear. They lack any identifiable cytoplasmic membrane or nuclear structure. They contain dense nuclear material and or chromatic strands. They are characteristic of chronic lymphocytic leukemia.
Hematologic malignancy characterized by progressive accumulation of phenotypically mature malignant B-cell lymphocytes in peripheral blood, bone marrow, and lymph nodes.
Rare, indolent B-cell lymphoproliferative disorder characterized by infiltration of bone marrow and spleen by neoplastic cells with cytoplasmic hair-like projections; considered a distinct form of mature B-cell neoplasm.
Low risk, characterized by lymphocytosis alone.

SLL is a different manifestation of the same disease as CLL, with the main difference being that abnormal lymphocytes mainly accumulate in lymph nodes and other tissues, with no evidence of cytopenias or bone marrow involvement. 1
Chronic lymphocytic leukemia
20-50% of patients
About 10%
About one-third of patients
BLACK BOX
No. Although all intravenous iron formulations can cause infusion reactions, their safety profiles differ. Older or less stable complexes are associated with a higher risk of hypersensitivity reactions, while newer carbohydrate shells have greatly improved tolerability.
Iron Dextran (INFeD) and Ferumoxytol (Feraheme) have black box warnings, largely because of past safety signals of serious hypersensitivity and anaphylaxis. The other IV iron formulations don’t carry boxed warnings because their risk profiles (both in clinical trials and postmarketing surveillance) have been more favorable, and no strong enough signal has required that level of regulatory warning. But the difference is not absolute — it reflects regulatory caution, historical risk, and observed reaction rates more than a mechanistic “safe vs unsafe” split.
- Iron Dextran (INFeD / older dextran forms)
- Dextran-based iron formulations historically had a higher rate of true anaphylactic (IgE-mediated) reactions, especially the older high–molecular-weight dextran.
- Because dextran itself is antigenic, even dextran-only infusions (without iron) had documented anaphylaxis cases historically.
- Low–molecular-weight iron dextran (INFeD) inherited the class label, so the warning stays: “Anaphylactic-type reactions, including fatalities, have been reported following parenteral administration of iron dextran.” FDA Access Data
- The black box is thus partly legacy, partly precaution based on historical risk.
- Ferumoxytol (Feraheme)
- After its introduction, a number of serious hypersensitivity and anaphylactic events were reported in postmarketing surveillance. U.S. Food and Drug Administration+2OptumRx Professionals+2
- The FDA responded by mandating a boxed warning indicating the risk of serious hypersensitivity / anaphylaxis and hypotension. FDA Access Data+1
- The label explicitly warns to administer slowly (≥15 minutes), monitor patients for at least 30 minutes after infusion, and have resuscitation equipment available. FDA Access Data+2FDA Access Data+2
Why other formulations don’t (yet) have black box warnings
- Their observed incidence of serious reactions has been much lower in both clinical trials and postmarketing data.
- No regulatory agency has judged that their risk justifies a boxed warning.
- Their formulations (non-dextran, more stable, smaller nanoparticle architectures) are considered inherently safer with lower likelihood of antigenic reaction or complement activation at typical infusion rates.
- For example:
- In a pharmacovigilance study, iron dextran and ferumoxytol had the highest reporting odds ratio (ROR) for hypersensitivity reactions. Iron sucrose and ferric carboxymaltose had lower RORs. PMC
- In comparative clinical use, ferumoxytol did not show a significantly higher rate of hypersensitivity/hypotension vs other compounds in some analyses, suggesting risk is low overall—but the boxed warning remains as a precaution
Key nuance & caveats
- Black box warning ≠ “unsafe” — it means extra caution is warranted, not that the drug must be avoided. Many drugs with serious risk carry boxed warnings yet are used widely under controlled conditions.
- Even in formulations without a black box, hypersensitivity or infusion reactions can still occur (though rarely).
- Regulatory labeling can lag behind mechanistic knowledge and real-world experience; a safe formulation today might acquire stronger warnings later if new signals emerge.
Summary
Iron dextran (INFeD) and ferumoxytol (Feraheme) carry boxed warnings for hypersensitivity and anaphylaxis due to historical and postmarketing safety signals. Other modern IV iron formulations lack boxed warnings because their observed rates of serious reactions have remained very low, though no IV iron is zero-risk. The absence of a black box does not imply absolute safety — rather, it reflects a balance of clinical trial data, postmarket surveillance, and regulatory judgment.
IRON TRIVIA
Yes, but it is extraordinarily rare—and almost never with modern formulations.
True, IgE- or IgG-mediated anaphylaxis was historically associated with the old high-molecular-weight iron dextran preparations (no longer marketed in the U.S.). Those reactions were due to the dextran shell, not the iron itself, and sometimes occurred even with dextran solutions that contained no iron.
With today’s formulations—low-molecular-weight iron dextran (INFeD®), ferumoxytol (Feraheme®), ferric carboxymaltose (Injectafer®), ferric derisomaltose (Monoferric®), ferric gluconate (Ferrlecit®), and iron sucrose (Venofer®)—the risk of anaphylaxis is less than 1 in 200,000 infusions, and fatalities are essentially unheard of. Most reactions that appear “allergic” are actually complement-activation–related pseudoallergies (CARPA) rather than true IgE-mediated events.
When in doubt, treat first and sort later: if a patient develops airway compromise, hypotension, or generalized urticaria, manage as anaphylaxis with epinephrine and supportive care, then review the event afterward to determine whether it was true allergy or CARPA.
Bottom line: Modern IV iron can rarely cause true anaphylaxis, but the likelihood is exceedingly low. Most apparent “allergic” reactions are non-allergic, complement-mediated events, and IV iron remains one of the safest parenteral treatments in clinical use.
Modern IV iron formulations are extremely safe.
Serious hypersensitivity reactions are exceedingly rare—estimated at fewer than 1 per 200,000 infusions, with no fatalities reported for current low-molecular-weight or non-dextran products. The discontinuation of high-molecular-weight iron dextran (the older, high-risk formulation) has dramatically improved the safety profile of IV iron therapy.
The vast majority of reactions seen today are mild, transient events—typically brief flushing, warmth, chest or back pressure, or anxiety (so-called Fishbane reactions). These occur in roughly 1–3% of infusions, resolve within minutes when the infusion is paused, and do not represent allergy.
Modern pharmacovigilance studies and meta-analyses confirm that IV iron is among the safest parenteral therapies used in clinical practice, with a risk of serious reaction comparable to or lower than that of many routine intravenous medications.
In short: With current formulations and proper infusion technique, IV iron is both safe and well-tolerated. Most patients complete treatment uneventfully, and true anaphylaxis is vanishingly rare.
Management depends on the severity of the reaction.
Most reactions to IV iron are mild, self-limited CARPA-type (“Fishbane”) events that resolve quickly when the infusion is paused. The key principle is to stop the infusion, observe, and restart slowly once symptoms abate.
1. Mild (Fishbane-type) reaction
- Typical features: Flushing, warmth, chest or back pressure, anxiety, mild dyspnea.
- Action:
- Stop the infusion immediately.
- Reassure the patient; monitor vital signs.
- Symptoms usually resolve within 5–10 minutes.
- Restart at half the previous rate once asymptomatic; most patients tolerate resumption without recurrence.
- Do not premedicate with antihistamines or steroids—they provide no benefit and may confuse the picture.
2. Moderate or uncertain reaction
- Features: Persistent symptoms, mild hypotension, or diagnostic uncertainty (Fishbane vs anaphylaxis).
- Action:
- Stop the infusion.
- Give oxygen and IV fluids as needed.
- Observe closely.
- If symptoms progress or airway involvement appears, treat as anaphylaxis.
3. Severe reaction (true anaphylaxis)
- Features: Hypotension, wheeze, stridor, urticaria, angioedema, collapse.
- Action:
- Stop infusion immediately.
- Administer intramuscular epinephrine (0.3–0.5 mg of 1:1000 solution).
- Provide airway and hemodynamic support (oxygen, IV fluids).
- Add antihistamines and corticosteroids only after epinephrine.
- Document and refer for allergy evaluation before any future IV iron exposure.
Follow-up
- Document the event clearly (timing, symptoms, vital signs, management, outcome).
- Distinguish between Fishbane-type and true anaphylaxis to avoid unnecessary “iron allergy” labeling.
- Most patients who experience mild or moderate CARPA-type reactions can safely receive IV iron again with slower infusion rates.
In summary: Pause, observe, and restart slowly for mild reactions; treat promptly with epinephrine for true anaphylaxis. Most IV iron reactions are mild, non-allergic, and do not preclude future use.
- Iron sucrose
- Sodium ferric gluconate complex
- Low molecular weight iron dextran
- Ferumoxytol
- Ferric carboxymaltose
- Ferric derisomaltose
Adverse reactions to modern IV iron are rare and usually mild. They can be grouped into three mechanistic categories:
1. Complement activation–related pseudoallergy (CARPA)
- The vast majority of reactions belong here.
- Mechanism: innate immune activation from nanoparticle–complement interaction, releasing anaphylatoxins (C3a, C5a).
- Clinical spectrum:
- Mild (“Fishbane reaction”) — transient flushing, warmth, back or chest pressure, anxiety; resolves quickly when infusion paused.
- Severe (non-Fishbane CARPA) — rarer; may include hypotension, dyspnea, or syncope; still non-IgE-mediated.
- Management: stop infusion, wait for resolution, restart at half rate; do not treat routinely with antihistamines or steroids.
2. True anaphylaxis (IgE- or IgG-mediated)
- Exceptionally uncommon (< 1 per 200,000 infusions).
- Historically associated with high-molecular-weight iron dextran (no longer marketed).
- Requires epinephrine; future exposure to same product contraindicated.
3. Other rare or delayed events
- Local infusion-site reactions (extravasation, discomfort).
- Delayed arthralgia/myalgia hours later (non-allergic, self-limited).
- Hypophosphatemia with certain formulations (esp. ferric carboxymaltose).
Summary:
Most infusion reactions to IV iron are CARPA-type events, with Fishbane reactions representing their mild, transient expression and non-Fishbane reactions their more severe form. True IgE-mediated anaphylaxis is vanishingly rare with today’s non-dextran formulations.
A Fishbane reaction is a mild, transient infusion reaction that occurs in roughly 1–3% of IV iron infusions, typically within the first few minutes of administration. It is not an allergic reaction but rather a benign form of complement activation–related pseudoallergy (CARPA) — an innate immune phenomenon triggered by nanoparticles of IV iron.
Mechanism
The reaction is thought to result from transient activation of the complement system when iron–carbohydrate complexes first enter the bloodstream. This leads to the release of anaphylatoxins (C3a, C5a), which briefly cause pulmonary vasoconstriction and stimulation of afferent autonomic pathways. The result is a sudden but short-lived sensation of pressure or tightness in the chest, back, or flanks, sometimes accompanied by flushing, warmth, anxiety, or tachycardia.
Unlike IgE-mediated allergy, no antibodies, mast cell priming, or sensitization are involved — and patients are not “allergic” to IV iron.
Clinical features
- Timing: Within 1–5 minutes of starting infusion
- Symptoms: Flushing, warmth, back or chest pressure, anxiety, sometimes mild dyspnea or tachycardia
- Duration: Usually resolves within 5–10 minutes after pausing infusion
- Severity: Mild, self-limited; does not progress to anaphylaxis
- Vital signs: May show transient tachycardia or mild hypotension, but no vascular collapse, urticaria, or bronchospasm
Management
- Stop the infusion immediately.
- Reassure the patient and observe until symptoms resolve (usually within minutes).
- Restart at half the rate once asymptomatic; most patients tolerate resumption without recurrence.
- No premedication with steroids or antihistamines is necessary and may obscure the diagnosis.
- Future infusions can proceed normally, though many clinicians begin the next infusion slowly out of caution.
Clinical significance
Fishbane reactions are not a contraindication to future IV iron use. They represent the mildest expression of CARPA, distinct from both severe CARPA reactions and true anaphylaxis. Recognizing this prevents unnecessary labeling of patients as “iron allergic” and avoids withholding appropriate therapy.
IV iron is preferred when oral iron fails, isn’t tolerated, or can’t be absorbed, or when rapid repletion is clinically necessary.
Common indications include:
- Intolerance to oral iron (e.g., gastrointestinal side effects, nonadherence).
- Malabsorption (e.g., celiac disease, inflammatory bowel disease, post–bariatric surgery).
- Inflammatory states with impaired intestinal iron uptake (e.g., CKD, heart failure, chronic inflammation).
- Ongoing blood loss exceeding oral replacement capacity (e.g., heavy menstrual bleeding, GI bleeding).
- Need for rapid correction (e.g., preoperative anemia, symptomatic iron deficiency, late pregnancy).
- Functional iron deficiency in patients receiving erythropoiesis-stimulating agents (CKD, oncology).
In brief:
Oral iron is first-line for most mild, uncomplicated cases. IV iron is appropriate when oral therapy doesn’t work, isn’t feasible, or isn’t fast enough.
Speed of infusion
- Although CARPA is influenced by infusion rate and formulation stability, the prevailing mechanistic model holds that complement activation is triggered by the nanoparticle form of the iron complex, rather than solely by “free iron” release.
- Modern IV iron agents are colloidal nanoparticles whose surface chemistry, size, and charge can provoke complement binding and activation (via classical, lectin, or alternative pathways).
- Rapid infusion increases the instantaneous nanoparticle exposure to complement proteins, increasing the likelihood of activation before immune regulatory clearance can intervene.
- The composition, molecular weight, charge, and structural integrity of the nanoparticle shell determine how the particle behaves in plasma and how likely it is to trigger complement activation (CARPA) or release labile iron.
- These properties vary between different formulation, this infusion time differs.
- For example, ferric carboxymaltose is very stable and can be indufed over 13-30 min whereas iron sucrose is less stable and is over 1-2 h.
- Loosely bound (labile) iron may amplify downstream inflammatory effects but is not considered the primary initiator of CARPA in current formulations. However, the difference in allowable infusion speeds between IV iron formulations primarily reflects differences in nanoparticle structure and stability, not free iron release.
- Note: Iron dextran is very stable but more immunogenic (dextran allergy risk) so is given over 1-6 h.
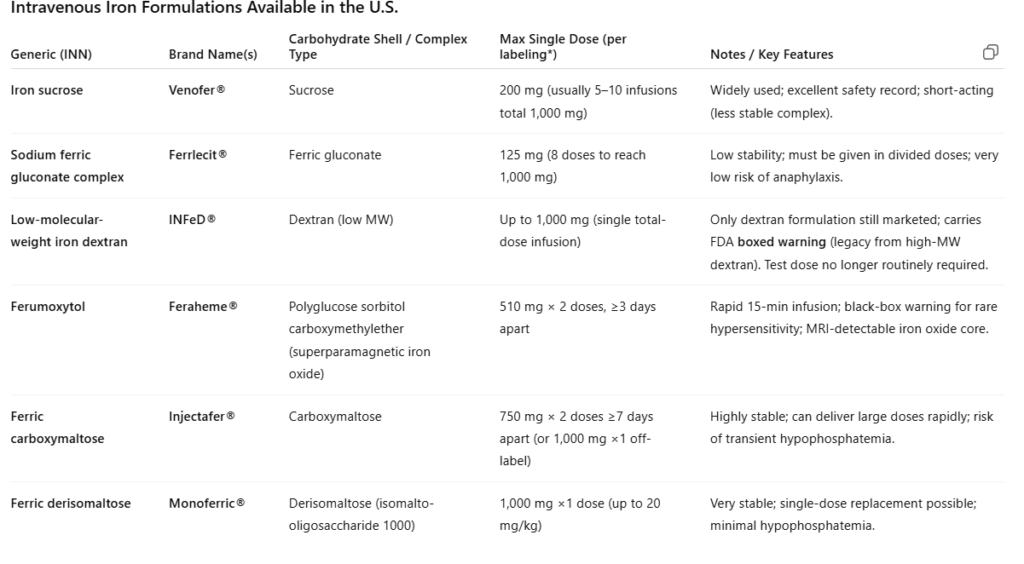
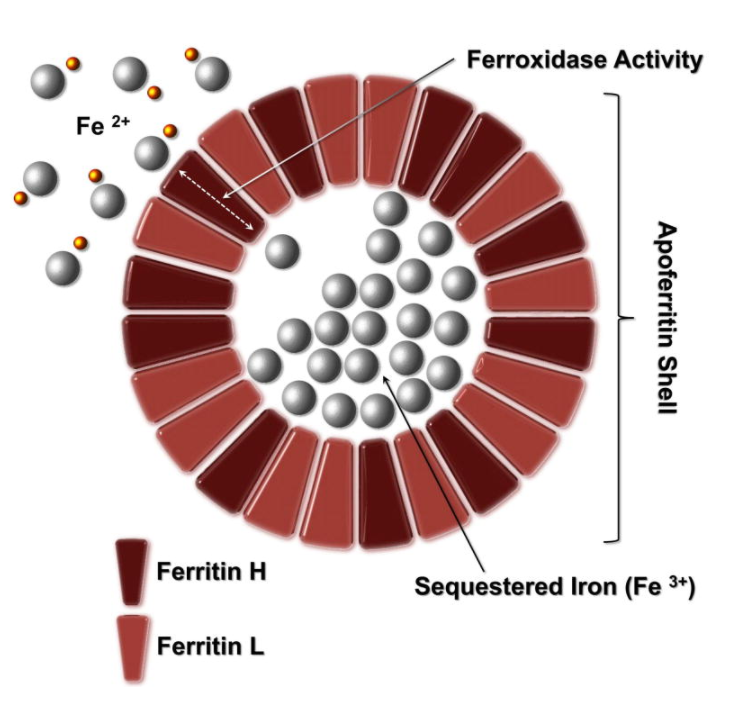
Ferritin Spike
After intravenous iron infusion, serum ferritin rises sharply within 24–48 hours, reflecting the large iron load taken up by macrophages and stored as ferritin. Transferrin saturation (TSAT) may increase modestly or remain normal, since only a fraction of infused iron is immediately released into plasma for binding to transferrin. Over subsequent days to weeks, ferritin gradually declines as stored iron is mobilized to transferrin and used for erythropoiesis.
After intravenous iron administration, serum ferritin often rises sharply for several days. This is not a reflection of iron overload but rather the expected physiologic handling of IV iron by the reticuloendothelial system:
- Iron uptake by macrophages:
IV iron complexes (e.g., iron dextran, ferric carboxymaltose) are taken up by macrophages of the liver, spleen, and bone marrow. - Intracellular ferritin synthesis:
Within macrophages, iron is stored in ferritin molecules — an immediate buffering response to the sudden influx of elemental iron. - Ferritin release into plasma:
Some ferritin is secreted into circulation, leading to a transient rise in serum ferritin levels.
Meanwhile, hepcidin increases, reducing iron export through ferroportin. As a result, transferrin saturation (TSAT) remains in the normal range despite the ferritin spike. Over the following weeks, iron is slowly released from macrophages and incorporated into erythropoiesis, and ferritin levels gradually decline toward a new steady state.

FISHBANE
Mechanistically, they’re on the same spectrum
- Both so-called Fishbane and non-Fishbane reactions:
- Occur within minutes of starting IV iron.
- Are non-IgE, complement-driven, innate immune events.
- Are rate-dependent and idiosyncratic.
- Share the same core mechanism: transient C3a/C5a generation leading to vasodilation, tachycardia, dyspnea, and anxiety.
- The only real difference is the magnitude of this activation and the extent of physiologic response.
So mechanistically, they are not two different entities—just different magnitudes of the same process. Same mechanism, different intensity
There’s no hard mechanistic line between a “Fishbane reaction” and a “non-Fishbane CARPA reaction.” The difference is operational, not biologic.
- Why we still label one as “Fishbane”:
- The term Fishbane reaction persists because it communicates prognosis and safety, not mechanism.
- Fishbane (mild CARPA)
- Brief, self-limited
- Stop, reassure, restart slowly
- Safe to rechallenge
- Non-Fishbane (moderate/severe CARPA)
- Hypotension, hypoxia, collapse
- Stop permanently, supportive care
- Avoid rechallenge; use alternative formulation
- At the bedside, that distinction answers the question “Can I safely restart this infusion?”
- That’s the true utility—not in mechanistic purity, but in risk stratification.
- But in clinical practice, the binary classification (mild vs severe) is more actionable than debating whether it’s “Fishbane” or “CARPA.”
- Fishbane (mild CARPA)
- The term Fishbane reaction persists because it communicates prognosis and safety, not mechanism.
- How most experts handle it in practice:
- At the bedside, most hematologists and infusion nurses now treat the terminology pragmatically:
- If symptoms are mild and resolve promptly → call it a Fishbane (mild infusion) reaction and restart slowly.
- If symptoms are more significant (hypotension, hypoxia, persistent distress) → call it hypersensitivity/infusion reaction (CARPA-like) and do not rechallenge.
- In either case, do not label the patient “allergic to IV iron” unless there’s evidence of true anaphylaxis.
- At the bedside, most hematologists and infusion nurses now treat the terminology pragmatically:
- Clinically, the distinction between Fishbane and non-Fishbane reactions is one of severity, not mechanism.
- Both are complement-mediated pseudoallergies; the “Fishbane” label simply signals the benign, self-limited end of that spectrum—important mainly to justify safe continuation of therapy.
- Fishbane and non-Fishbane CARPA reactions share a common complement-mediated mechanism. The only clinically meaningful distinction is severity — the presence of hypotension, hypoxia, or collapse transforms a benign, self-limited Fishbane episode into a more serious infusion reaction requiring discontinuation
- Clinically convenient distinction:
- “Fishbane” → safe to restart once symptoms resolve.
- “Severe CARPA” → stop permanently, switch formulation.
- That’s really the bedside decision point.
FAQ About IV Iron
- Serious hypersensitivity reactions to modern IV iron formulations are exceedingly rare. Large pharmacovigilance studies estimate anaphylaxis in fewer than 1 per 200,000 infusions, with no fatalities reported for current low-molecular-weight or non-dextran preparations.
- Mild, transient reactions (such as flushing or chest tightness—so-called Fishbane reactions) occur in about 1–3% of infusions and usually resolve within minutes when the infusion is paused.
- The discontinuation of high-molecular-weight iron dextran has made IV iron one of the safest parenteral therapies used in clinical practice.
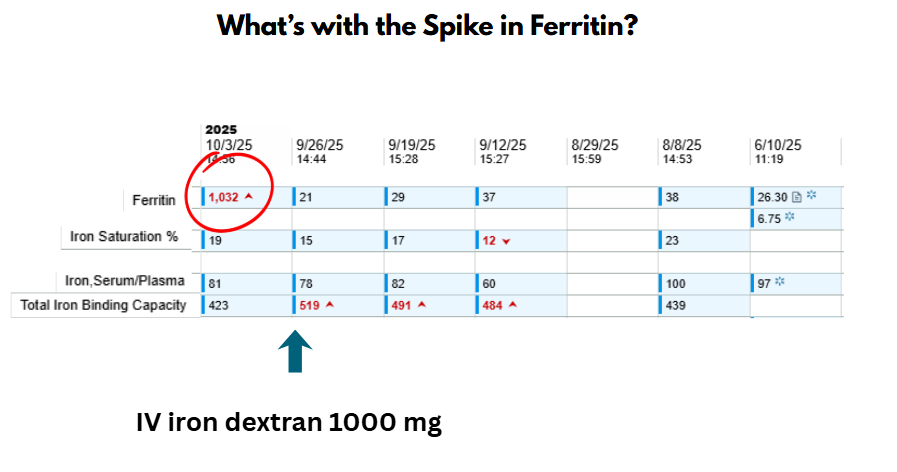
Mild (Fishbane / minor CARPA):
- Stop infusion → Observe → If symptoms resolve in ≤10 min, restart at slower rate
- Step-by-Step Management:
- Stop the infusion.
- Observe — symptoms typically resolve within 5–10 min.
- When completely resolved:
- Restart infusion at ≤50% rate.
- Monitor closely.
- If symptoms recur → stop and do not restart.
- Do not give premedication for future infusions; it’s not preventive and can mask early warning signs.
Moderate CARPA:
- Stop infusion → Monitor → Give IV/PO antihistamine ± corticosteroid → Resume only if full resolution
- Step-by-Step Management:
- Stop the infusion.
- Assess vitals; provide oxygen if needed.
- Administer:
- Diphenhydramine 25–50 mg IV or PO, or second-generation antihistamine if mild.
- Methylprednisolone 40–80 mg IV if persistent symptoms or recurrent reaction.
- Resume infusion only if symptoms completely resolve.
- Document event and consider switching formulation for future treatment.
Severe (Anaphylaxis):
- Stop infusion → Epinephrine immediately → Supportive care (O₂, fluids) → Emergency transfer
- Step-by-Step Management:
- Stop infusion immediately.
- Call for emergency assistance (code/EMS).
- Administer epinephrine promptly:
- 0.3–0.5 mg (0.3–0.5 mL of 1:1000) IM in mid-thigh; Repeat every 5–10 min if no improvement.
- Lay patient supine, elevate legs unless contraindicated.
- Oxygen 8–10 L/min via mask.
- IV fluids (normal saline or lactated Ringer’s) rapidly.
- Adjuncts:
- Antihistamine (diphenhydramine 25–50 mg IV)
- Corticosteroid (methylprednisolone 125 mg IV)
- Bronchodilator if wheezing (albuterol neb)
- Monitor continuously; prepare for airway management if needed.
- Transfer to emergency department for observation ≥4–6 h (risk of biphasic reaction).
It Depends on the Formulation
- Each IV iron product has its own maximum infusion rate and allowable single dose, determined by the stability of the iron–carbohydrate complex.
- Infusion speed is limited not by “toxicity” per se, but by the risk of transient complement activation (Fishbane-type or CARPA reactions) that correlate with how fast free nanoparticles enter the circulation.
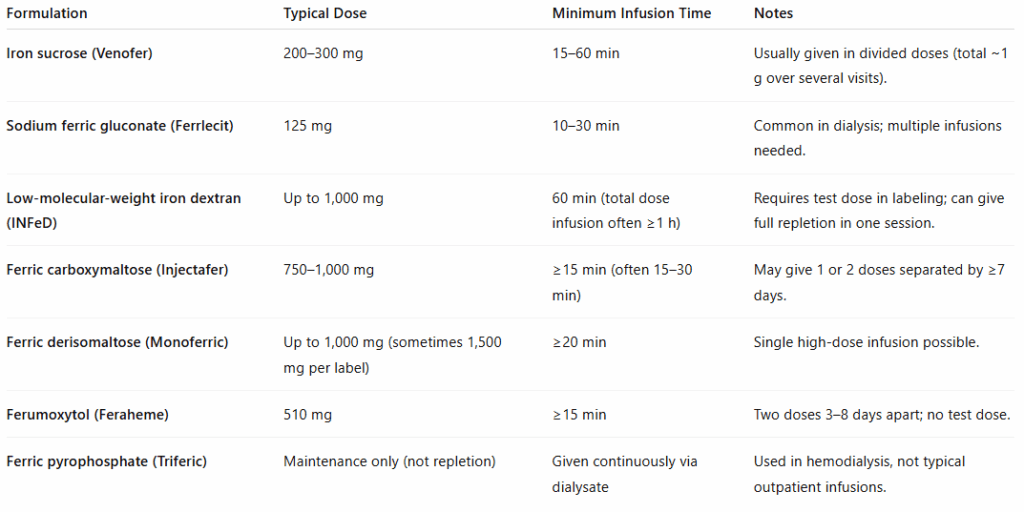
Key Takeaways
- Infusion rate is formulation-specific — always follow the product’s label.
- 15–60 minutes is the general range for modern IV irons.
- Observation after infusion is standard for safety.
- Slower rates can reduce benign pseudoallergic (CARPA/Fishbane) reactions.
If two patients receive IV iron from the same bag and at the same rate, will both have a reaction?
No. These reactions are largely patient-dependent, not product-dependent.
- A complement-activation reaction (sometimes called a Fishbane reaction) reflects an individual’s innate sensitivity to nanoparticle iron complexes, which can vary widely between patients.
- Factors such as baseline complement reactivity, underlying inflammation, genetic differences, or even anxiety and catecholamine tone can influence who reacts.
- So if patient A develops flushing or chest tightness, it doesn’t mean there’s anything wrong with the bag or that patient B will react too.
- Each infusion is essentially a new “test” of the patient’s own physiology — not of the product itself.
- Urticaria is a clinical pattern, not a mechanism. It simply means mast-cell–mediated wheals and pruritus, which can arise from:
- IgE cross-linking (true allergy),
- Complement activation (CARPA) releasing C3a, C5a (anaphylatoxins),
- Direct mast-cell degranulation via non-IgE triggers (opioids, contrast, IV iron nanoparticles).
- Urticaria during IV iron infusion ≠ automatically IgE-mediated.
- Most urticarial reactions are non–IgE complement-mediated pseudoallergies (CARPA).2
- True IgE-mediated urticaria/anaphylaxis is exceedingly rare and would require prior sensitization and confirmatory testing.
- True IgE-mediated urticaria would generally:
- Occur after prior exposure to that same formulation,
- Appear rapidly and reproducibly on re-challenge,
- Possibly progress to anaphylaxis if untreated,
- Show positive skin or in vitro testing (rarely done).
- In practice, we treat based on severity, not on presumed mechanism — but understanding this distinction helps avoid overlabeling patients as “iron allergic.”
- Otherwise Healthy Adults With Iron Deficiency Anemia (e.g., From Menorrhagia):
- Evidence Summary:
- Several RCTs compare IV versus oral iron in women with iron-deficiency anemia from menstrual or GI blood loss.
- Findings:
- IV iron (sucrose, carboxymaltose) produces faster hemoglobin and ferritin recovery and earlier symptom relief.
- Oral iron remains effective but limited by GI intolerance and slower correction.
- Interpretation:
- In straightforward IDA, IV iron is not first-line, but is appropriate when oral therapy fails, is intolerable, or rapid restoration is needed (e.g., pre-op or severe menorrhagia).
- Evidence Summary:
- Non-Anemic Iron Deficiency:
- Evidence Summary:
- Small RCTs in non-anemic individuals with low ferritin, often women with fatigue or chronic symptoms.
- Findings:
- IV iron can reduce fatigue scores and improve well-being when ferritin <50 µg/L.
- No consistent improvement in objective performance or cognition.
- Response varies by baseline iron status and symptom burden.
- Interpretation:
- IV iron may be symptomatically helpful in select NAID patients, but evidence is heterogeneous; use should be individualized and generally after oral trial.
- Evidence Summary:
- Elite Athletes:
- Evidence Summary:
- Small physiologic RCTs in endurance athletes with low ferritin or training at altitude.
- Findings:
- IV (or IM) iron increases ferritin and TSAT, sometimes improving VO₂max and training adaptation.
- Performance gains are inconsistent and small.
- Greater benefit seen in female and altitude-exposed athletes.
- Interpretation:
- IV iron can be useful for iron-deficient athletes when oral therapy fails or time is limited, but routine use is not recommended; must follow anti-doping (WADA) infusion limits.
- Evidence Summary:
- Chronic Kidney Disease (CKD):
- Evidence Summary:
- CKD is the most extensively studied setting for IV iron, both in dialysis-dependent and non-dialysis patients. Trials have compared IV versus oral iron and tested different IV formulations and dosing strategies.
- Findings:
- IV iron consistently raises hemoglobin faster and more reliably than oral iron.
- Reduces erythropoiesis-stimulating agent (ESA) requirements.
- Major RCTs (e.g., DRIVE, FIND-CKD, PIVOTAL) show safety when ferritin and TSAT are appropriately monitored.
- Infection and cardiovascular risk remain low with current protocols.
- Interpretation:
- IV iron is standard of care in CKD-associated anemia, particularly when oral therapy is ineffective or rapid correction is desired.
- Evidence level: Strongest and most mature across all populations.
- Evidence Summary:
- Heart Failure:
- Evidence Summary:
- RCTs have evaluated IV iron in patients with heart failure and iron deficiency, with or without anemia.
- Findings:
- Trials such as FAIR-HF, CONFIRM-HF, AFFIRM-AHF, and IRONMAN show improved functional status, exercise tolerance, and quality of life.
- Some trials demonstrate fewer heart-failure–related hospitalizations.
- Mortality impact remains less certain.
- Interpretation:
- IV iron (especially ferric carboxymaltose or ferric derisomaltose) is recommended in HFrEF with iron deficiency to improve symptoms and reduce rehospitalization risk.
- Evidence level: High, reflected in guideline recommendations (ACC/AHA, ESC).
- Evidence Summary:
- Pregnancy / Postpartum:
- Evidence Summary:
- Multiple RCTs compare IV versus oral iron for iron deficiency anemia during pregnancy or postpartum.
- Findings:
- IV iron (iron sucrose, ferric carboxymaltose) yields faster hemoglobin recovery, higher ferritin, and better tolerance.
- Decreases fatigue and improves maternal well-being.
- No increase in adverse obstetric or neonatal outcomes.
- Interpretation:
- IV iron is safe and effective in the second and third trimesters or postpartum when oral iron fails, is not tolerated, or rapid repletion is necessary.
- Evidence level: Moderate to high, depending on formulation.
- Evidence Summary:
- Perioperative / Surgical:
- Evidence Summary:
- IV iron has been tested as preoperative optimization therapy in anemic or iron-deficient surgical patients.
- Findings:
- IV iron raises preoperative hemoglobin and iron stores.
- Evidence for reducing transfusion or improving outcomes is mixed, particularly if given <1–2 weeks before surgery.
- Works best when administered early and combined with erythropoietin in select cases.
- Interpretation:
- IV iron is useful preoperatively in iron-deficient patients when there’s adequate lead time; benefit on hard outcomes varies.
- Evidence level: Moderate, supportive for early preoperative use in iron-deficient patients.
- Evidence Summary:
- Inflammatory Bowel Disease (IBD):
- Evidence Summary:
- Several RCTs compare IV versus oral iron in Crohn’s disease and ulcerative colitis.
- Findings:
- IV iron (iron sucrose, ferric carboxymaltose) corrects anemia more effectively and with fewer gastrointestinal side effects than oral formulations.
- Improves fatigue and quality of life.
- Interpretation:
- IV iron is preferred in active IBD or when oral iron causes GI intolerance or mucosal irritation.
- Evidence level: Moderate to high.
- Evidence Summary:
- Oncology / Chemotherapy-Induced Anemia:
- Evidence Summary:
- RCTs evaluate IV iron alone or in combination with ESA therapy in patients receiving chemotherapy.
- Findings:
- IV iron increases hemoglobin response and reduces ESA requirements.
- Modest improvement in quality of life; no clear survival impact.
- Safe across cancer types.
- Interpretation:
- IV iron is reasonable adjunctive therapy for chemotherapy-induced anemia, particularly in ESA-treated patients with low iron indices.
- Evidence level: Moderate, but often limited by heterogeneity of cancer type and concurrent therapies.
- Evidence Summary:
- Chronic Inflammatory / Rheumatologic Disorders:
- Evidence Summary:
- Smaller RCTs in rheumatoid arthritis and other inflammatory diseases.
- Findings:
- IV iron improves hemoglobin and fatigue modestly.
- Safety profile similar to that in IBD and CKD.
- Interpretation:
- IV iron can be considered in chronic inflammation–associated anemia when functional iron deficiency is present.
- Evidence level: Low to moderate.
- Evidence Summary:
- Miscellaneous / Other Populations:
- Heart and lung transplant candidates, restless legs syndrome, bariatric surgery patients, and heart failure with preserved EF (HFpEF) — emerging or pilot RCTs suggest benefit but are not yet guideline-strength.
- Critically ill / ICU patients: Mixed results; ongoing trials.
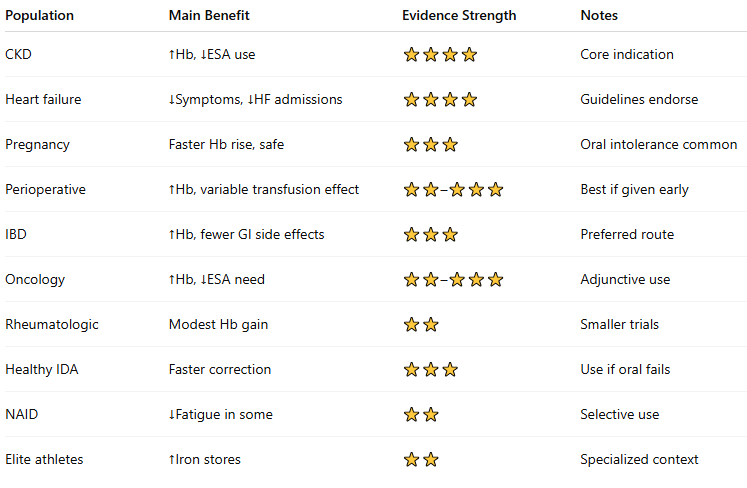
- IgE-mediated (true anaphylaxis)
- Complement activation–related pseudoallergy (CARPA)
- Fishbane reaction
- Non_Fishbane CARPA
- Delayed (non-immediate) hypersensitivity
Hypersensitivity reactions” to IV iron refer to acute, systemic adverse events that occur during or shortly after infusion, usually within minutes. They range from mild, self-limited symptoms to life-threatening anaphylaxis.
Historically, these reactions were most common with high-molecular-weight iron dextran (no longer marketed in the U.S.) but can occur — rarely — with any formulation.
Modern preparations (iron sucrose, ferric carboxymaltose, ferumoxytol, ferric derisomaltose, etc.) have a very low rate of serious hypersensitivity (<1 per 200,000 doses for anaphylaxis).
IV iron is used when oral iron is ineffective, not tolerated, or too slow to correct deficiency.
Typical situations include:
- Malabsorption (e.g., celiac disease, inflammatory bowel disease, bariatric surgery)
- Ongoing blood loss exceeding oral absorption (e.g., heavy menstrual bleeding, GI bleeding)
- Inflammatory states or CKD, where hepcidin blocks intestinal iron uptake
- Intolerance to oral iron (nausea, constipation, poor adherence)
- Need for rapid repletion, such as preoperative anemia, late pregnancy, or severe deficiency with symptoms
In short, IV iron is chosen when speed, absorption, or tolerance make oral therapy impractical.
- Because most acute reactions to IV iron are complement-activation (non-IgE) events, not true allergic reactions.
- When the infusion is stopped, complement activation rapidly subsides and symptoms (flushing, chest tightness, myalgia) usually resolve within minutes.
- Since there’s no ongoing immune sensitization, the infusion can often be restarted slowly from the same bag once the patient is symptom-free and stable, typically at half the previous rate with close monitoring.
- This would never be done after a true anaphylactic (IgE-mediated) reaction, where the drug must be permanently discontinued.
If a patient has a mild reaction to IV iron, isn’t the problem in the bag? Shouldn’t I change bags or lots before restarting?
- No. These mild reactions are not caused by contamination or a bad bag, but by a transient immune response in the patient—specifically, complement activation triggered by nanoparticles of iron entering the circulation too quickly.
- Once the infusion is stopped, this reaction rapidly settles, and restarting slowly from the same bag is safe because the drug itself isn’t “faulty.”
- Changing the bag or lot won’t prevent the reaction; what matters is the infusion rate and careful monitoring.
- If the reaction were allergic (IgE-mediated) or severe, then the infusion would be permanently stopped and not restarted at all—regardless of bag or lot.
Because it can worsen the situation or mask the diagnosis.
- For many years, clinicians routinely gave diphenhydramine as premedication or treatment for IV iron reactions — borrowing habits from transfusion or contrast medicine.
- However, updated understanding of IV iron hypersensitivity (especially the CARPA mechanism) has changed this practice.
- Most acute IV iron reactions are complement activation–related pseudoallergies, not histamine-driven allergic reactions.
- Thus, antihistamines like diphenhydramine don’t address the underlying cause and may make clinical assessment more difficult.

When (and How) It May Be Used
Diphenhydramine is reasonable only in:
- Moderate allergic-type reactions with clear urticaria, itching, or angioedema after IV iron (especially if mild and non-progressive).
- Adjunctive therapy after epinephrine in anaphylaxis — never as first-line.
If used:
- Give 25–50 mg IV or PO after stabilization, not preemptively.
- Avoid routine premedication.
Bottom Line
Diphenhydramine is not contraindicated, but:
- It is no longer first-line for IV iron reactions.
- It should not be given preemptively or early in hypotensive reactions.
- It may be used selectively for urticaria after stabilization.
Tutorial: Hemolysis
Yes. Haptoglobin is an acute-phase reactant and can be falsely elevated in inflammation, masking the consumption from hemolysis. In addition, very mild or intermittent hemolysis may not overwhelm haptoglobin clearance.
Yes. In “compensated hemolysis,” the bone marrow increases red blood cell production enough to keep hemoglobin normal despite ongoing hemolysis.
Yes. Clostridial sepsis (via α-toxin), Mycoplasma pneumoniae (cold agglutinins), and Babesia (intraerythrocytic parasite) can all cause hemolysis through direct RBC destruction or immune mechanisms.
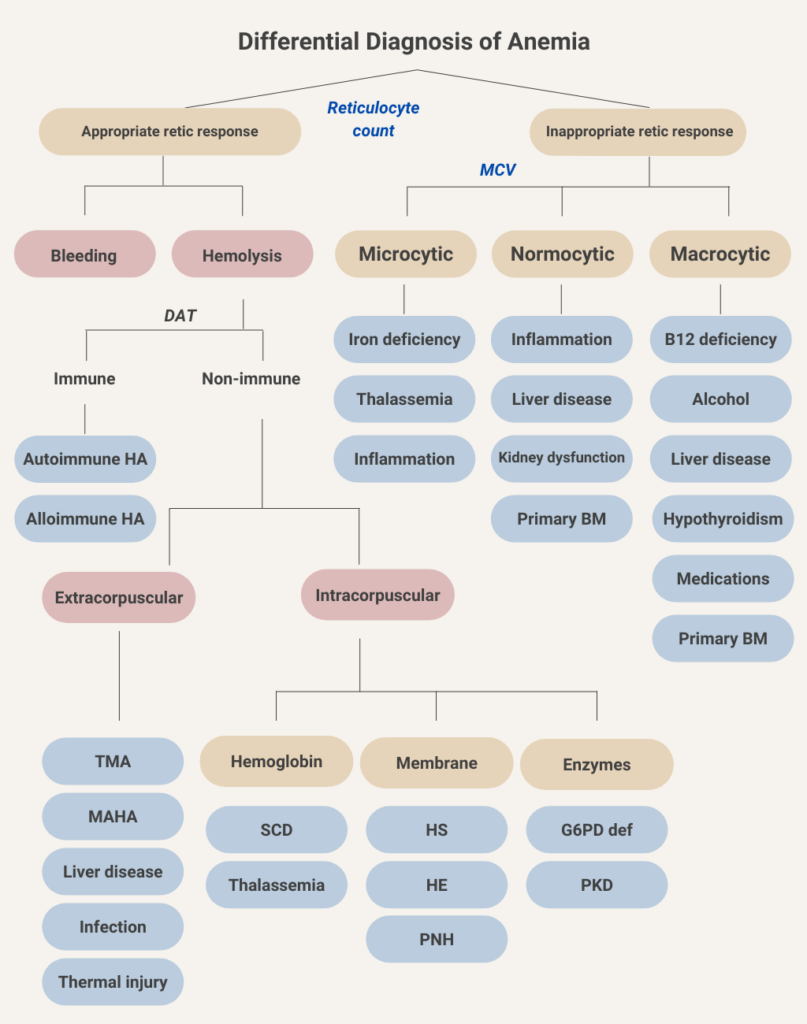
The diagnosis is based on a combination of:
- Clinical suspicion (e.g., anemia, jaundice, dark urine)
- Laboratory markers (LDH, haptoglobin, bilirubin, reticulocyte count)
- Exclusion of mimics
- Additional testing (DAT, peripheral smear, genetics, etc.) to determine the cause.
Malaria parasites rupture RBCs during their asexual cycle (direct destruction). Additionally, infected and uninfected RBCs are cleared by splenic macrophages due to altered membrane properties and immune recognition.
Common markers include:
- ↑ LDH (especially intravascular hemolysis)
- ↓ Haptoglobin
- ↑ Indirect bilirubin
- ↑ Reticulocyte count
- Hemoglobinuria/hemosiderinuria (in intravascular cases)
Cirrhosis, severe vitamin B12 deficiency, and resolving hematomas can all cause elevated indirect bilirubin or LDH that may resemble hemolysis.
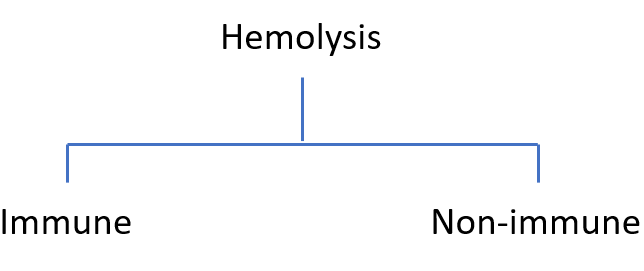
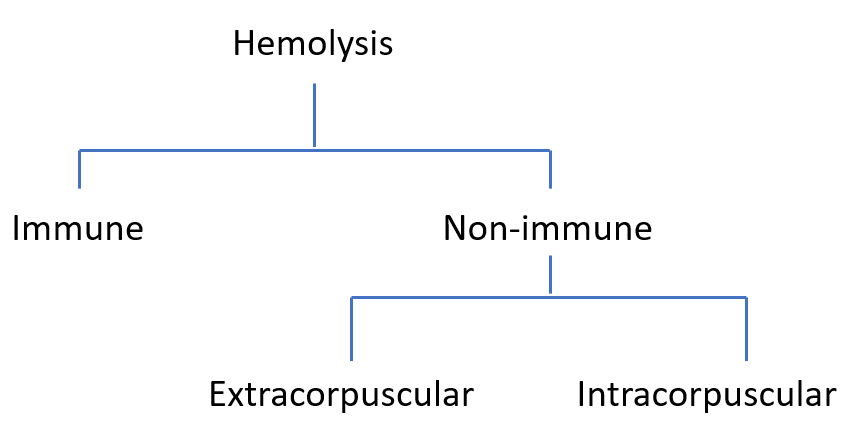
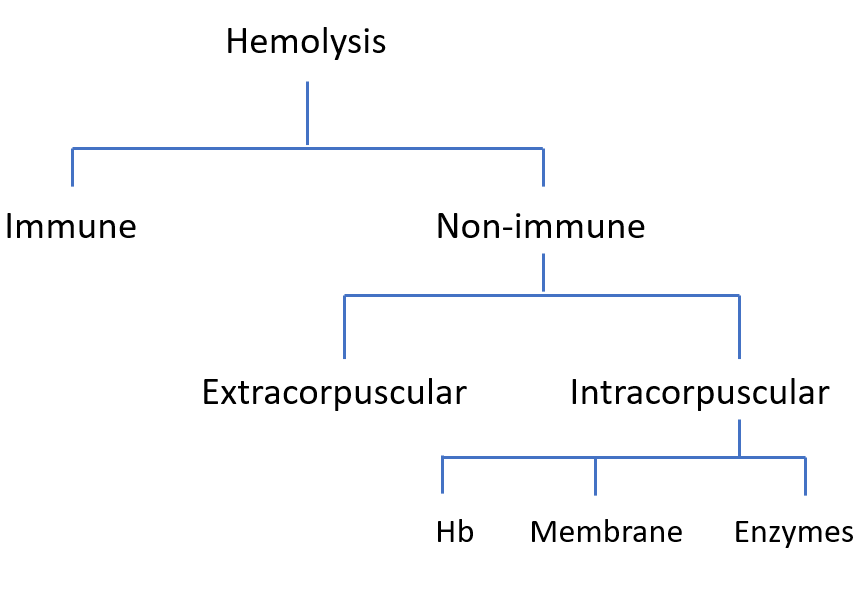
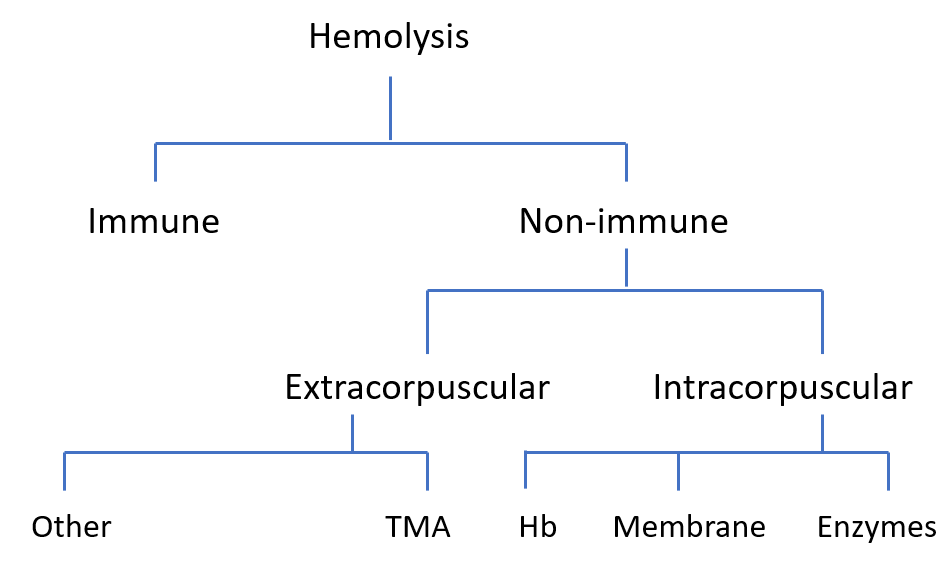
Hemolysis is the premature destruction of red blood cells. It can occur inside blood vessels (intravascular) or in the spleen and liver (extravascular).
The DAT detects immunoglobulin and/or complement coating the RBC surface. A positive test supports immune-mediated hemolysis (e.g., warm or cold autoimmune hemolytic anemia), while a negative DAT suggests non-immune causes.
- Hemolysis: reticulocytosis, low haptoglobin, hemoglobinuria (if intravascular).
- Cirrhosis: may raise indirect bilirubin but typically without low haptoglobin or reticulocytosis.
- B12 deficiency: can elevate LDH and indirect bilirubin due to intramedullary RBC death, but smear shows macrocytosis and hypersegmented neutrophils.
The liver has a high capacity to conjugate bilirubin. In chronic or slowly progressive hemolysis, hepatic clearance can keep pace, preventing hyperbilirubinemia despite ongoing RBC destruction.
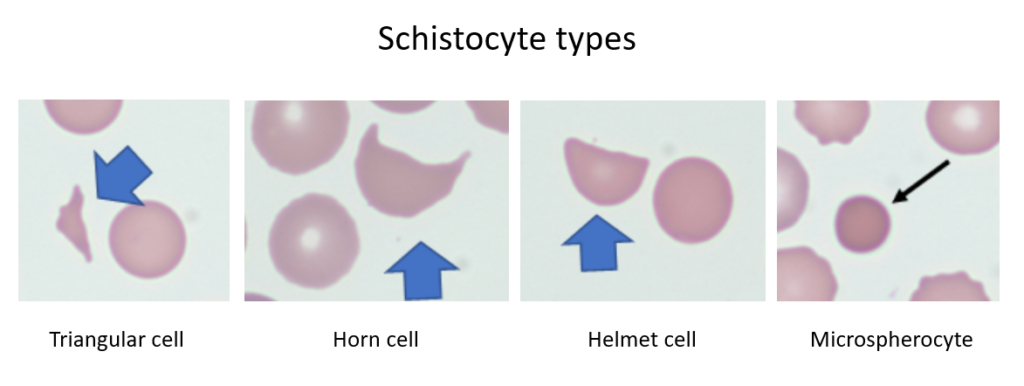
Narrow, fibrin-rich microvascular channels slice RBCs as they pass, producing fragmented cells (schistocytes) and intravascular hemolysis.
Free hemoglobin is released directly into plasma when RBCs lyse intravascularly. Once haptoglobin is saturated, unbound hemoglobin dimers are filtered by the kidney, appearing in urine. In extravascular hemolysis, intact RBCs are phagocytosed, so hemoglobin is not released directly into circulation.
Red cells are physically traumatized as they pass through turbulent flow or strike the prosthetic surface. This shear stress fragments cells, producing schistocytes and causing intravascular hemolysis.
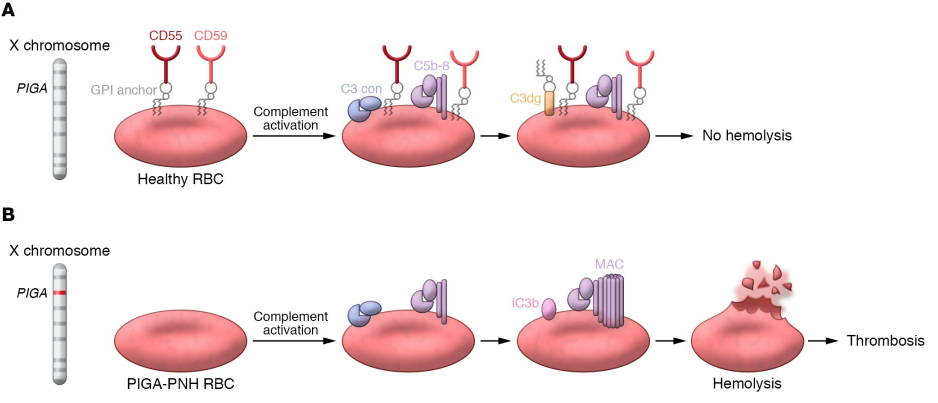
PNH RBCs lack GPI-anchored proteins that protect against complement. This makes them highly susceptible to complement-mediated lysis in circulation, leading to chronic intravascular hemolysis, hemoglobinuria, and risk of thrombosis.
Severe B12 deficiency leads to ineffective erythropoiesis (intramedullary destruction of RBC precursors). This raises LDH and indirect bilirubin, mimicking hemolysis, but haptoglobin is usually normal and the smear shows macrocytosis with hypersegmented neutrophils.
In intravascular hemolysis, red cells rupture directly in circulation, releasing large amounts of cytosolic LDH into plasma. In hereditary spherocytosis (extravascular), intact RBCs are engulfed and digested by macrophages, so LDH remains contained within the macrophage and is not markedly elevated.
Infections generate oxidative stress through activated neutrophils and cytokines. In G6PD deficiency, RBCs cannot regenerate NADPH effectively, so oxidant damage accumulates, leading to hemolysis.
Red blood cells
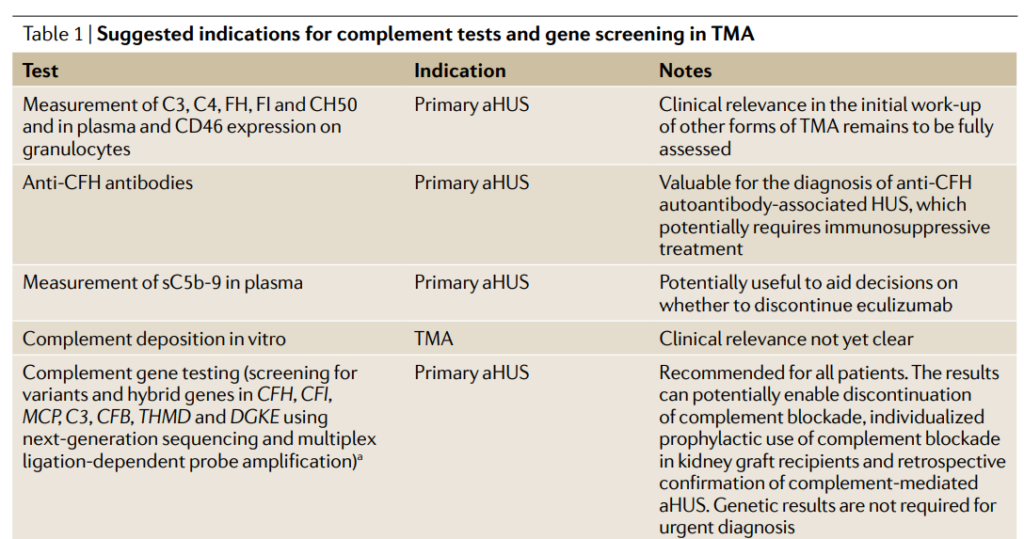
No. Normal complement concentrations do not exclude aHUS because low levels of circulating C3 have low sensitivity (about 30% of patients with aHUS), whereas high concentrations of circulating C5a and soluble C5b-9 might have insufficient specificity.
Learn more here.
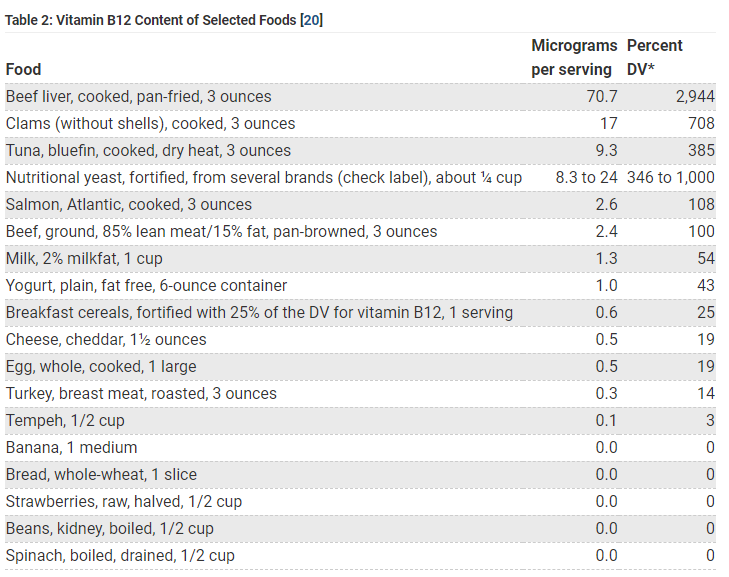
Yes, vitamin B12 is synthesized solely by microorganisms (ruminants obtain Cbl from the foregut) and is naturally present in foods of animal origin, including fish, meat, poultry, eggs, and dairy products. In addition, fortified breakfast cereals and fortified nutritional yeasts are readily available sources of vitamin B12 that have high bioavailability. The bioavailability of vitamin B12 is about three times higher in dairy products than in meat, fish, and poultry, and the bioavailability of vitamin B12 from dietary supplements is about 50% higher than that from food sources. Vegans are at increased risk for developing vitamin B12 deficiency. Read more here.
Yes. In some centers, the DAT (direct antiglobulin test) has largely replaced the term Coombs test. Coombs test was named after Robin Coombs, a British immunologist who developed the principle behind the antiglobulin test while travelling back to Cambridge on a wartime train.
Yes. Learn more here.
Acute attacks are very rare before puberty and after menopause, with a peak occurrence within the third decade. They are more common in women than in men. Most patients have one or a few attacks and then recover fully for the rest of their lives.
Learn more here.
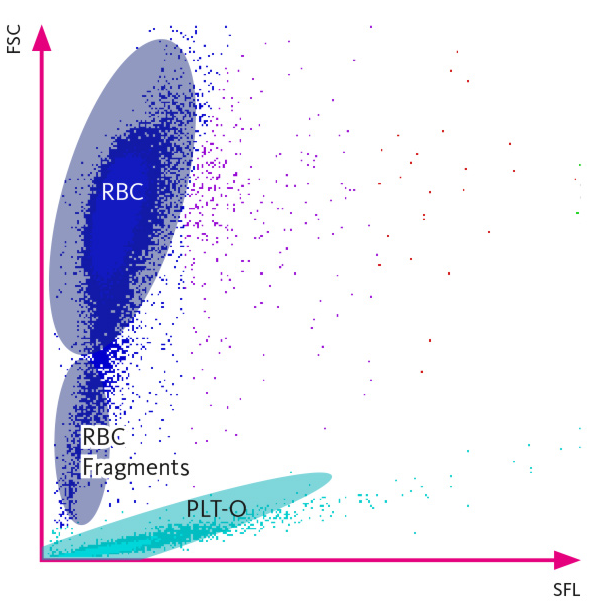
Fragmented RBCs can be detected by certain automated hematology analyzers based on the analysis of fraction of small red blood cells (RBCs) in the context of normal RBC volume indices (mean corpuscular volume and width). Their presence should prompt a peripheral blood smear review.

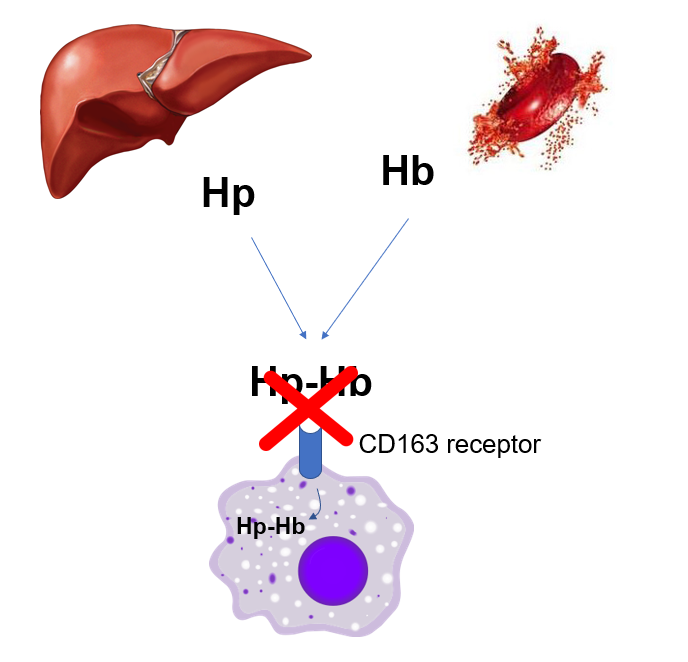
Rarely, haptoglobin (Hp) levels are normal or increased in hemolysis owing to inhibition of the macrophage uptake of the haptoglobin-hemoglobin complex (Hp-Hb) by the CD163 receptor.
Yes! AABB states that a single unit RBC transfusion should be standard in nonbleeding, hospitalized patients and additional units prescribed only after reassessment of patient and hemoglobin (Hb) level.
Food and Drug Administration (FDA) regulations permit hemochromatosis patients to donate blood, provided the donor (the hemochromatosis subject) meets standard blood donor eligibility criteria.
The American Red Cross does not permit hemochromatosis patients to donate blood because it “has a long-standing policy that potential donors are not allowed to receive direct compensation for their donation (beyond the usual orange juice and cookie). Because people with hemochromatosis would otherwise have to pay for their therapeutic phlebotomies, they would in effect be getting something of value for being able to donate for free. Thus the Red Cross has ruled that such donations violate their policy” (read more here).
Bottom line: blood-collection organizations can determine their own protocols within U.S. Food and Drug Administration regulations, which stipulate acceptable iron levels in donated blood.
In a 2016 Editorial, West and Eder wrote:
Safety concerns historically stem from the possibility of creating an incentive to donate blood for free rather than to pay for therapeutic phlebotomy, possibly encouraging HH donors to deny risk factors for infectious diseases. In the Final Rule that became effective in May 2016, the US Food and Drug Administration codified the requirements for hereditary hemochromatosis (HH) donors in the Code of Federal Regulations (CFR), thus eliminating the need for a variance to collect whole blood more frequently than every 8 weeks (or double red blood cells more frequently than every 16 weeks) and distribute units without special labeling from HH donors who meet all eligibility requirements. Notably, the CFR retains the requirement for obtaining a prescription for therapeutic phlebotomy from a licensed health care provider and performing therapeutic phlebotomy free of charge… Since the molecular basis of the disease was elucidated in 1996, it has been posited that the condition itself poses no harm to the recipient. The question concerns the motives of the HH donor to give blood and the possibility of incentive to withhold information from blood establishments about infectious risk factors
In 2016, the American Red Cross wrote (despite its continued refusal to allow patients with HH to donate blood) a piece titled “Iron-rich blood is just fine, thank you!”:
For decades, blood centers in the United States would not collect whole blood from donors/patients with hereditary hemochromatosis (HH), in some cases because it used to be that such units had to be labeled with the disease necessitating its removal… In 2016, the FDA encoded the regulations for therapeutic phlebotomy… Special labeling is not required, and units may be distributed if they meet regular requirements and criteria, as long as the therapeutic phlebotomy (TP) is ordered by a physician and the phlebotomy performed without charge.
Rarely, owing to skewed X chromosome inactivation.
Thrombocytopenia is absent at presentation in 15–20% of patients.
Learn more here.
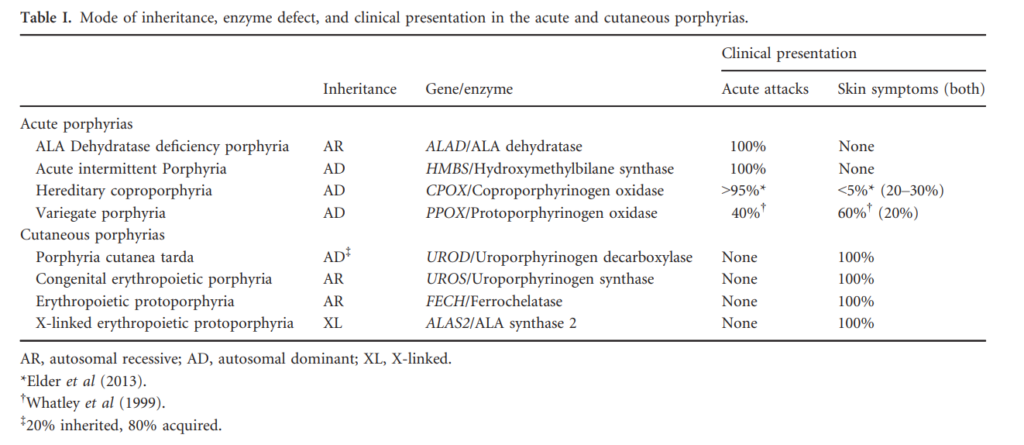
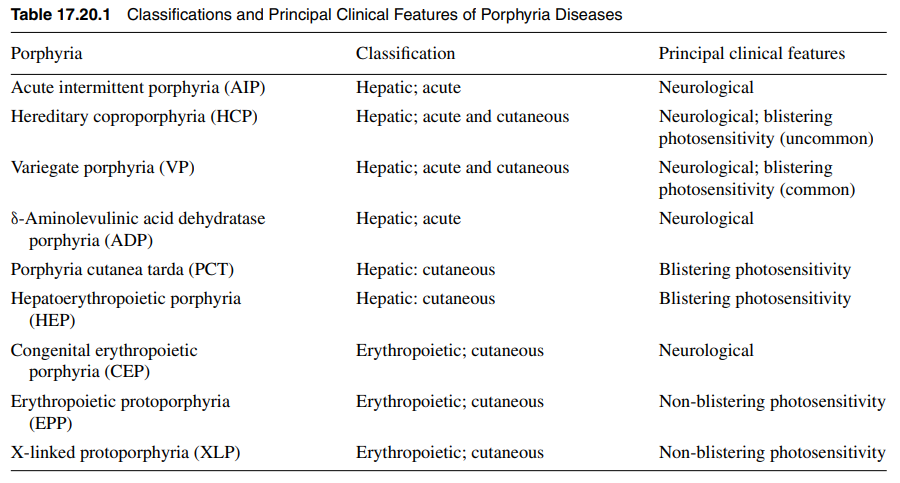
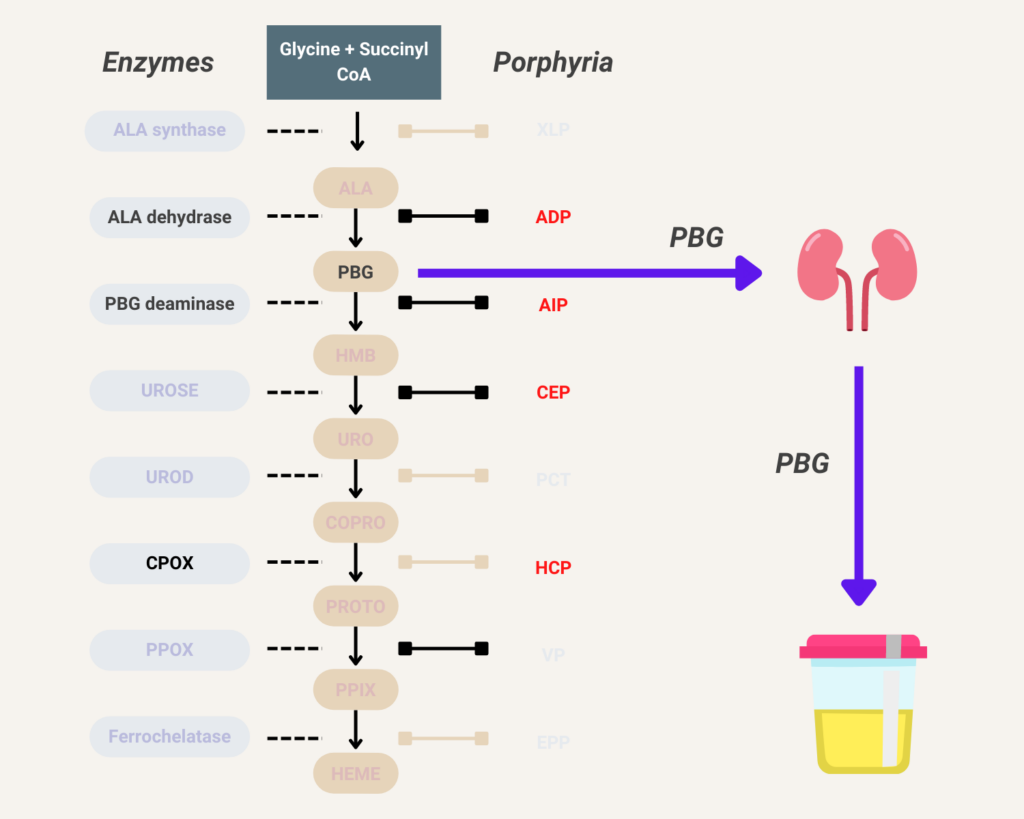
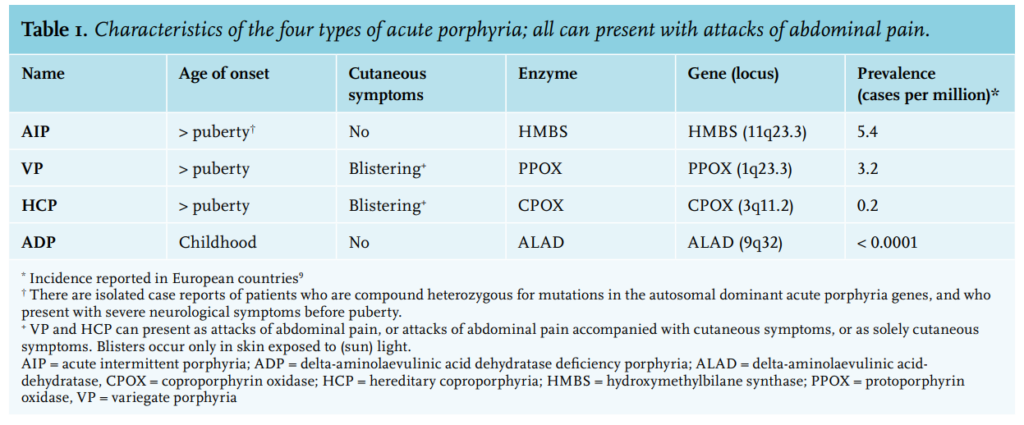
Skin lesions never develop in acute intermittent porphyria, but are the only clinical manifestation in some patients with variegate porphyria (60% of patients), and rarely (5%) develop in patients with hereditary coprophorphyria. Learn more here.
Yes, G6PD seems to be protective, as are many of the hemoglobinopathies and ethnic neutropenia. Learn more about hemoglobinopathies and malarial infection here.
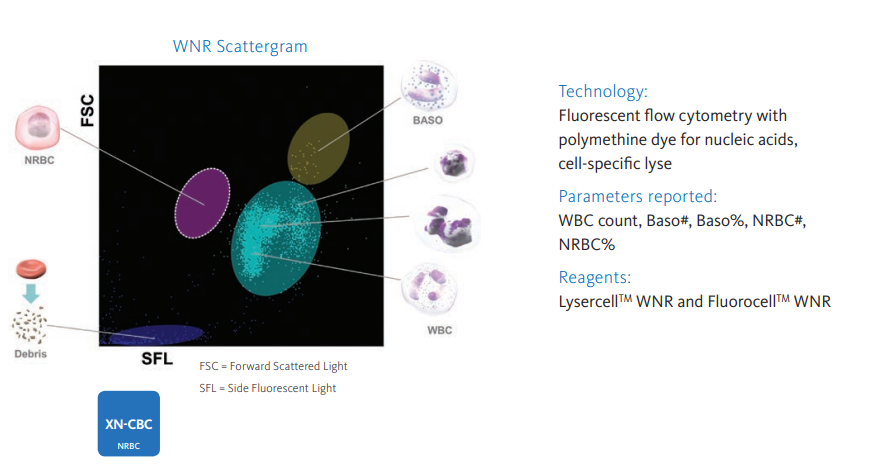
nRBCs are typically enumerated by one of the following methods:
- Manual counting of Wright-Giemsa-stained peripheral smears:
- Part of the traditional 100-cell manual differential leukocyte count.
- Reported as number of nRBCs per 100 white blood cells (WBCs).
- Automated hematology analyzers (for example, Sysmex)
- Reported as relative number of nRBCs per 100 WBCs or as an absolute number.

1) Reticulocyte stain:
The traditional method of measuring the reticulocyte count is a manual method that uses supravital stains (such as methylene blue) to highlight the reticulum (RNA) network of this immature red cell fraction. A lab technologist uses a microscope to count the number of such cells relative to the number of mature red blood cells. The number of reticulocytes is reported as a percentage of total red blood cells.
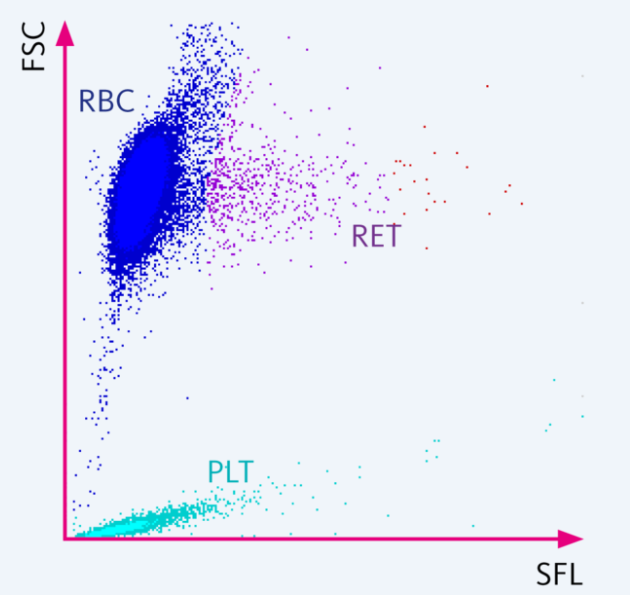
2) Automated analyzer:
The number of reticulocytes can be measured directly by most automated analyzers by staining the remnant RNA with a fluorescent dye. The number of reticulocytes is reported as an absolute count.
About one-half of patients with sickle cell disease have chronic ophthalmologic complications.
30%-70% of patients develop side effects, including:
- Nausea
- Vomiting
- Diarrhea
- Constipation
- Epigastric pain
- Metallic taste
Anemia occurs in about 75% of patients with chronic liver disease.
Learn more here.
Prevalence is 25-30%.
Prevalence is 10%-40% in adults with sickle cell disease (less common in children).
Thrombocytosis occurs in about 10-15% of patients with iron deficiency anemia. Learn more here.
Absolute vitamin B12 deficiency occurs in up to 6% of those aged 60 years and older. Learn more here.
If Hct > 54%, either reduce or stop testosterone therapy or initiate phlebotomy while continuing testosterone therapy.
Let’s look at the clinical practice guidelines:
2017 British Society for Sexual Medicine Guidelines on Adult Testosterone Deficiency, With Statements for UK Practice:

2018 Evaluation and Management of Testosterone Deficiency: AUA Guideline:

Testosterone Therapy in Men With Hypogonadism: An Endocrine Society Clinical Practice Guideline:

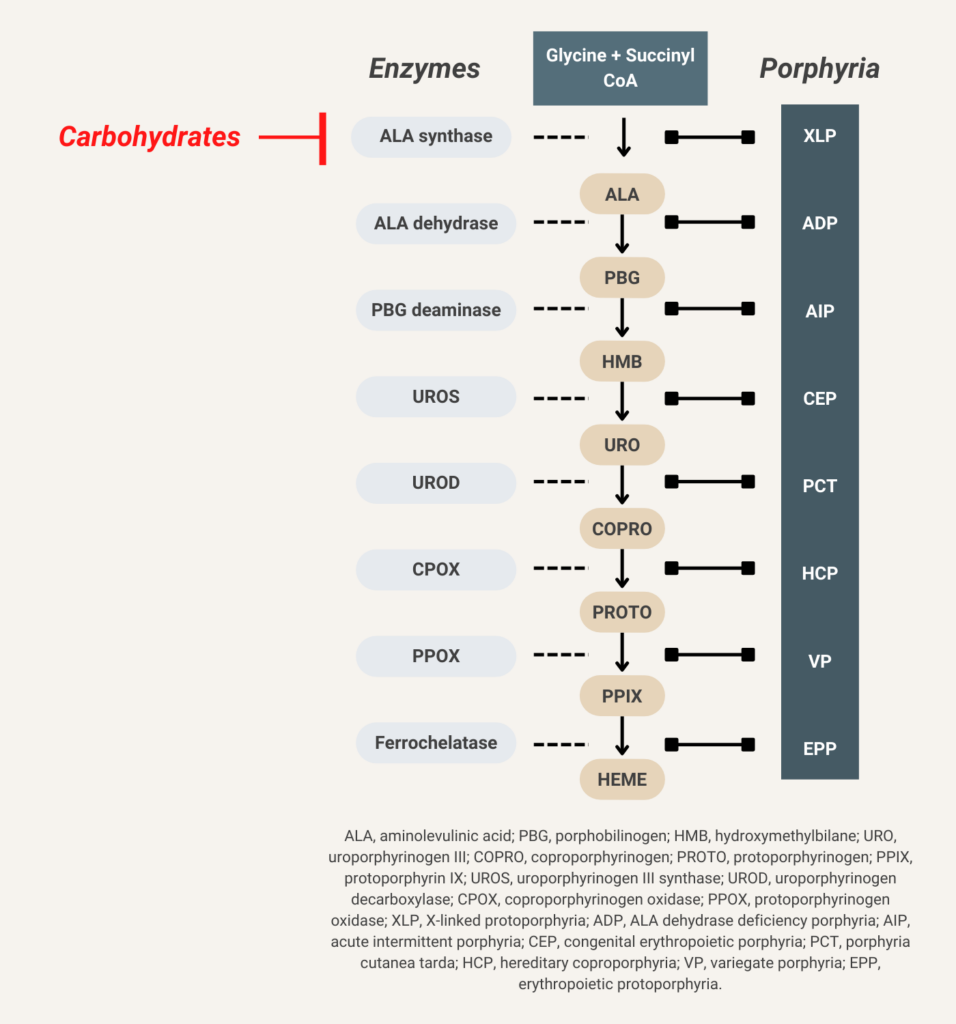
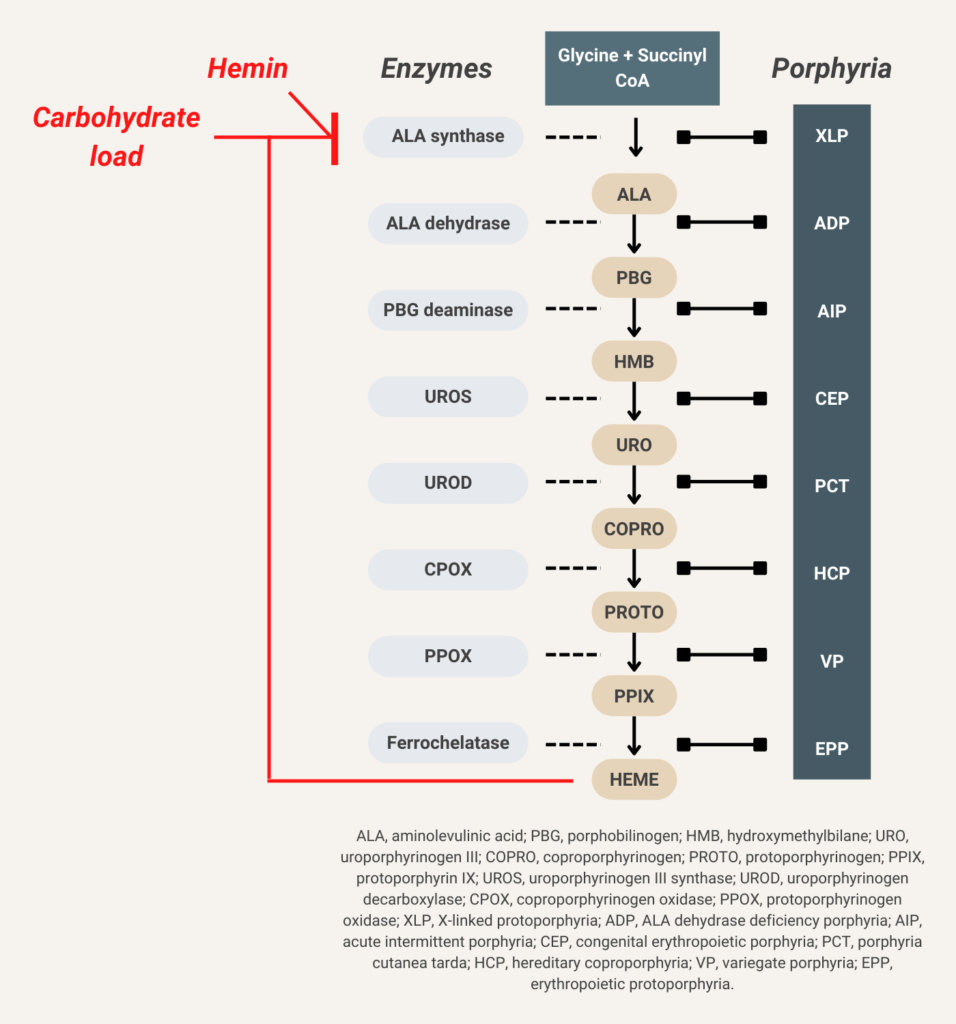
Glucose indirectly inhibits aminolevulinic acid (ALA) synthase activity and thereby decreases overproduction of ALA and porphobilinogen (PBG).
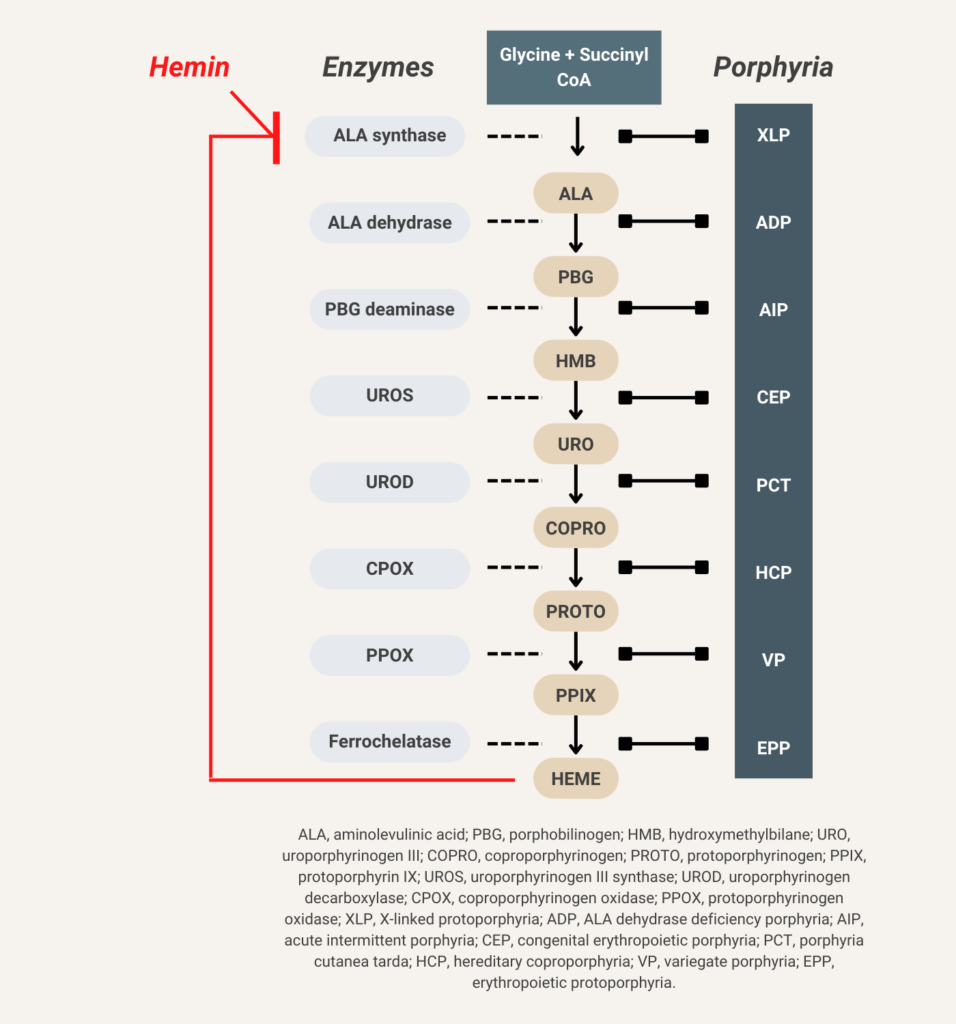
Hemin (available in the United States as Panhematin) suppresses aminolevulinic acid (ALA) synthase activity and thereby decreases overproduction of ALA and porphobilinogen (PBG).
Leukocyte reduction filters are used to remove > 99.9% of leukocytes. According to a Circular of Information from the AABB, American Red Cross, America’s Blood Centers, and the Armed Services Blood Program, leukocyte-reduced units of red blood cells must have a residual content of leukocytes <5.0 x 106.
About 70% of patients with pernicious anemia have macrocytosis. Learn more here.
Small clinically silent PNH clones are found in up to 70% of adults. Learn more here.
Reduction in hemoglobin ≥ 2 g/dL (20 g/L) below baseline (per NIH 2014 clinical practice guideline).
Two main classification schemes:
- Based on reticulocyte count (hypoproliferative vs. hyperproliferative)
- Based on red cell morphology (microcytic [with or without hypochromia]) vs. normocytic vs. macrocytic)
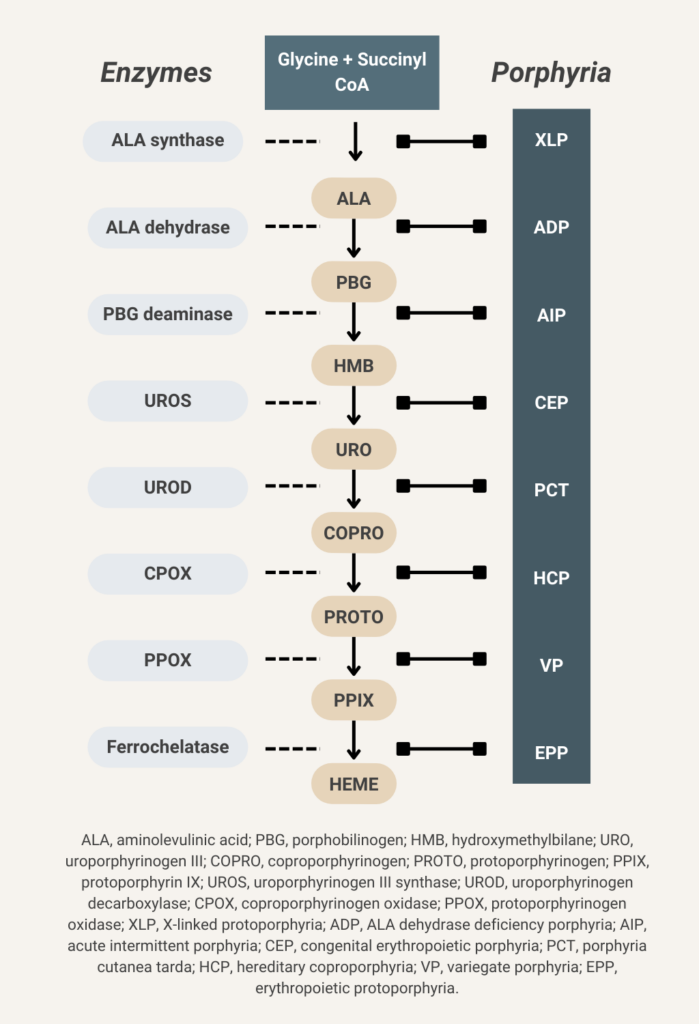
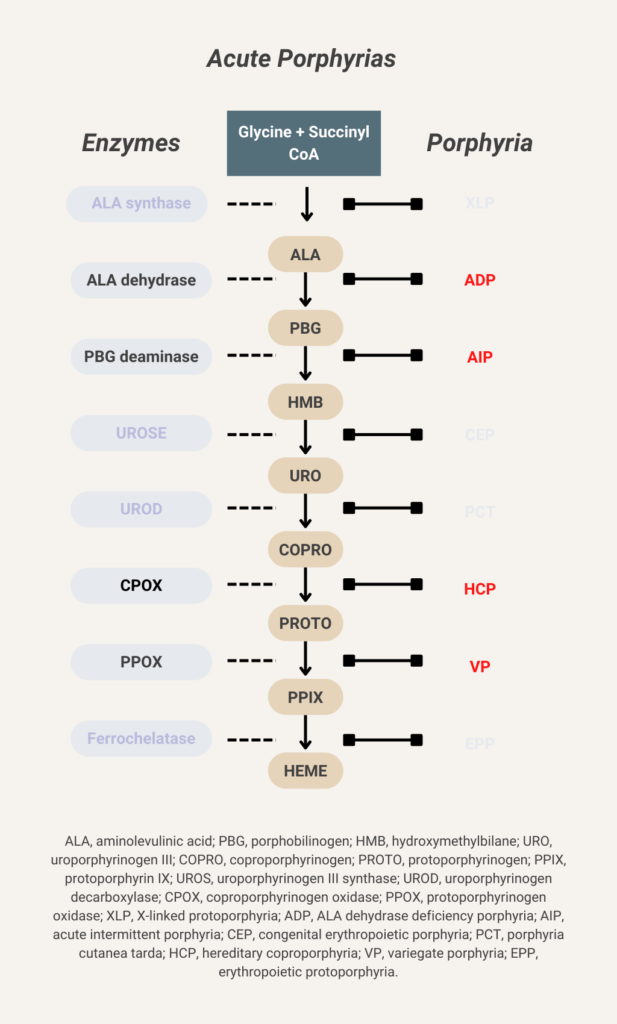
Elevated urine porphobilinogen (PBG) confirms diagnosis in acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), or variegate porphyria (VP). PBG level is normal in the very rare ALA dehydratase deficiency porphyria (ADP). Discriminating between AIP, HCP, and VP requires additional testing.
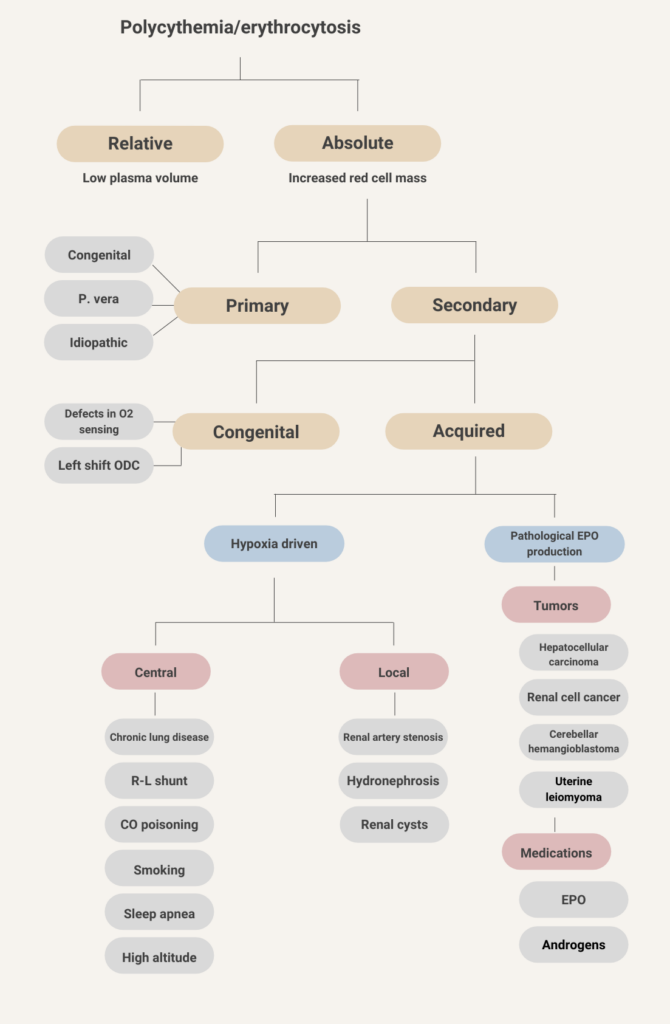
- Absolute erythrocytosis (elevated red blood cell mass)
- Primary – cause is intrinsic to the red blood cell
- Secondary – cause is extrinsic to the red blood cell
- Apparent or relative erythrocytosis (reduced plasma volume relative to red blood cell mass).
Learn more here.
Urine dipstick positive for blood + urine microscopy negative for red cells is consistent with hemoglobinuria or myoglobinuria. Differentiating between hemoglobinuria and myoglobinuria is usually obvious based on the clinical context (for example, serum hemolytic indices positive in hemoglobinuria and serum CK elevated in myoglobinuria), but definitive diagnosis can be made using mass spectrometry.
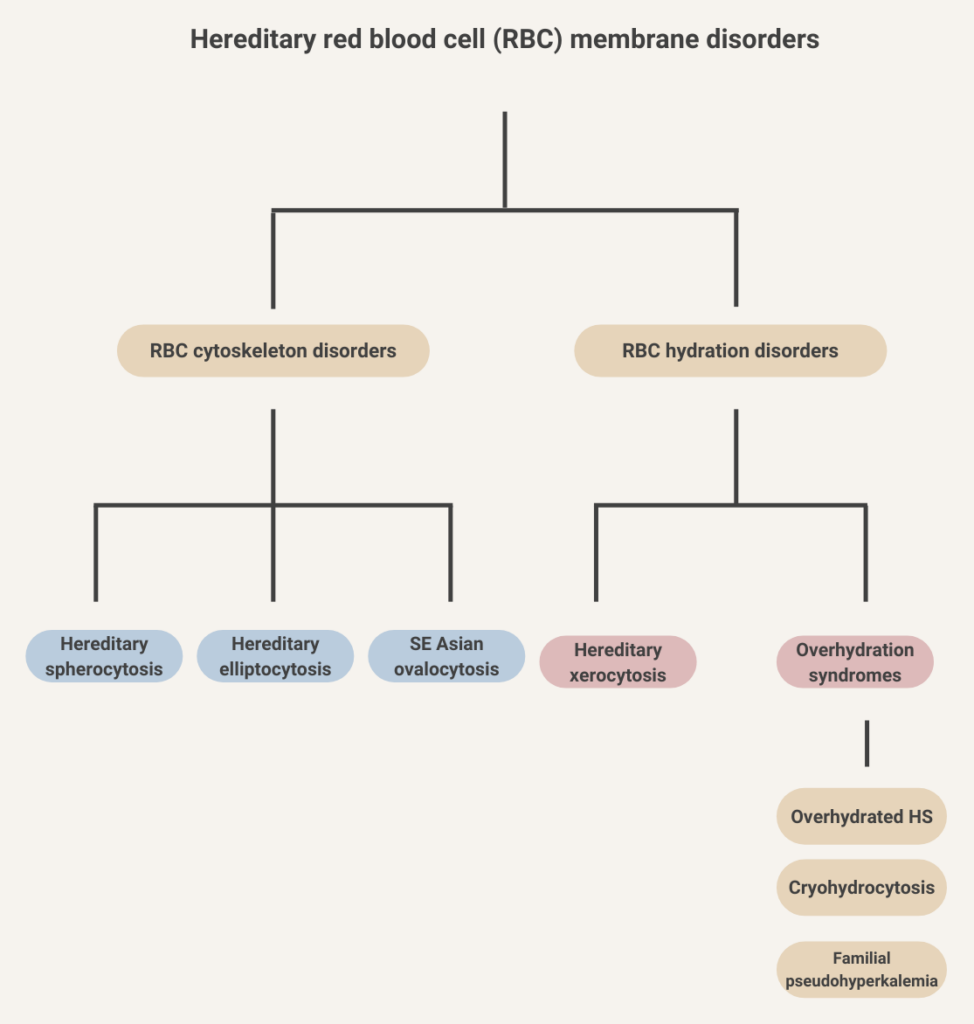
- Presence of elliptocytes on peripheral blood smear (fragmented red blood cells may also be seen).
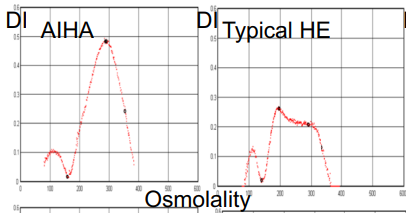
- Increased osmotic fragility.
- Osmotic gradient ektacytometry shows characteristic deformability profiles with curve exhibiting a trapezoidal form with a decrease in the RBC deformability.
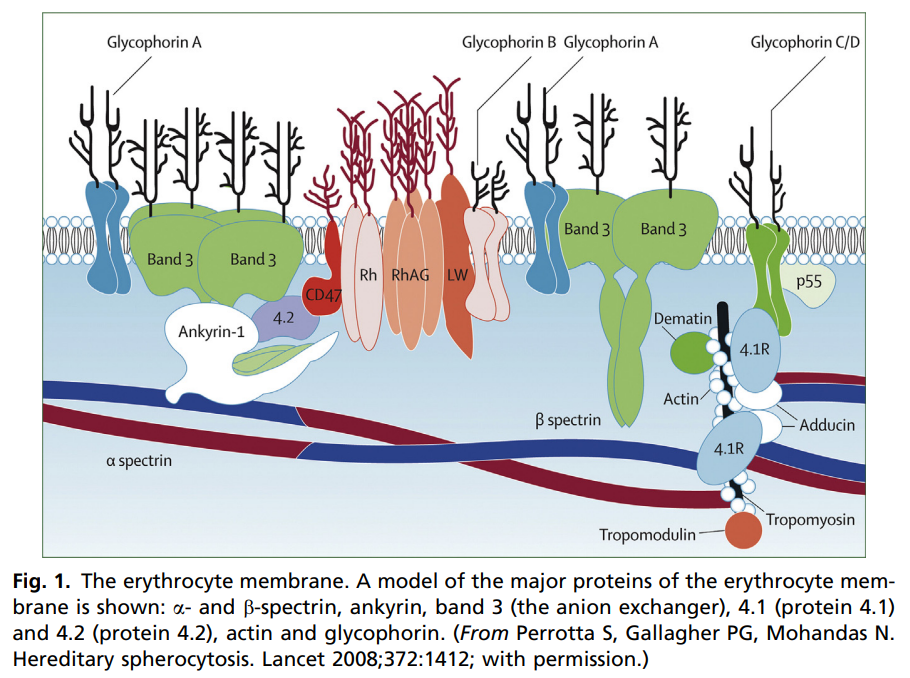
- DNA testing panels can define the pathogenic mutations in alpha-spectrin, beta-spectrin, and protein 4.1 in HE.
International Council for Standardization in Haematology (ICSH) guidelines one laboratory diagnosis of nonimmune hereditary red cell membrane disorders:

Learn more here.
- Presence of spherocytes on peripheral blood smear
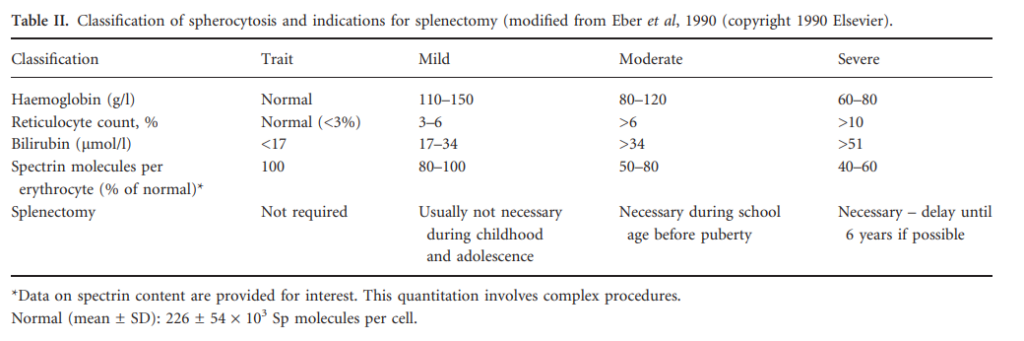
- Increased mean corpuscular hemoglobin concentration (MCHC) (> 36 g/dL [360 g/L])
- Increased lysis in osmotic fragility test and reduced fluorescence signal in eosin-5-maleimide (EMA) binding test
Diagnosis of HS does not necessarily require molecular analysis of affected genes.
2012 British Committee Standards in Haematology (BCSH) expert guideline on diagnosis of hereditary spherocytosis recommendations:


Flow cytometry using ≥ 2 different monoclonal antibodies against 2 different glycosylphosphatidylinositol (GPI)-anchored proteins (GPI-APs) on ≥ 2 different blood cell lineages. Learn more here.
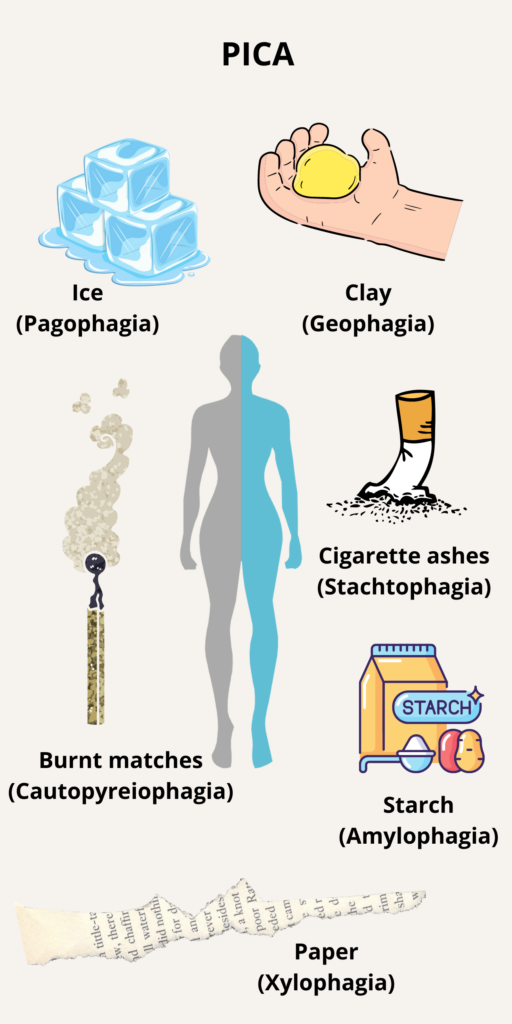
Pica is defined as the compulsive eating of non-nutritive substances. Learn more here.
- Primary (idiopathic)
- Secondary – associated with presence of other disorders, including iron deficiency
Learn more here.
By dividing the hematocrit (Hct) by the red blood cell count (RBC):
MCV = Hct/RBC
For example:
45% = 5 x 1012/L x 90 x 10-15L
0.45=450×10-3
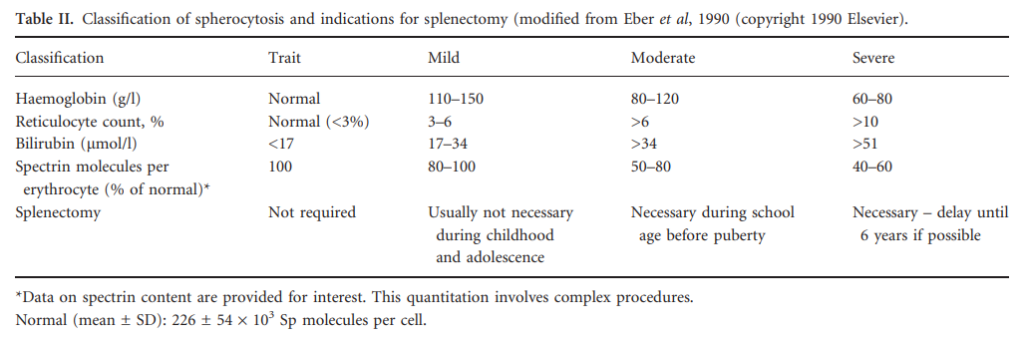
- Mild HS
- Normal Hb level with reticulocytes < 6%.
- May require 0-1 transfusion during lifetime and rarely splenectomy.
- Moderate HS
- Hb level > 8 g/dL (80 g/L) with reticulocytes 6%-10%.
- May require 0-2 transfusions during infancy and in some cases splenectomy (if the capacity level is decreased).
- Moderately severe HS
- Hb level 6-8 g/dL (60-80 g/L) with reticulocytes > 10%.
- May require > 2 transfusions intermittently and splenectomy likely necessary.
- Severe HS
- Hb level < 6 g/dL (60 g/L) with reticulocytes > 10%.
- Require regular transfusions and splenectomy likely necessary.
2012 British Committee Standards in Haematology (BCSH) expert guideline on diagnosis of hereditary spherocytosis:
Learn more here.
- 21 days if stored in citrate phosphate dextrose (CPD), or CPD2 anticoagulant-preservative
- 35 days if stored in citrate phosphate dextrose adenine 1 (CPDA-1)
- 42 days using current generation of additive solutions additive solutions
Most acute attacks last for no longer than 1 or 2 weeks.
Learn more here.
Continue oral iron for 3-6 months after the iron deficiency has been corrected in order to replenish iron stores.
About 4,500
> 1000! Learn more here.
100 million per year worldwide, 13 million in the US.
About 2 x 1011 (200 billion) or 1% of all red cells
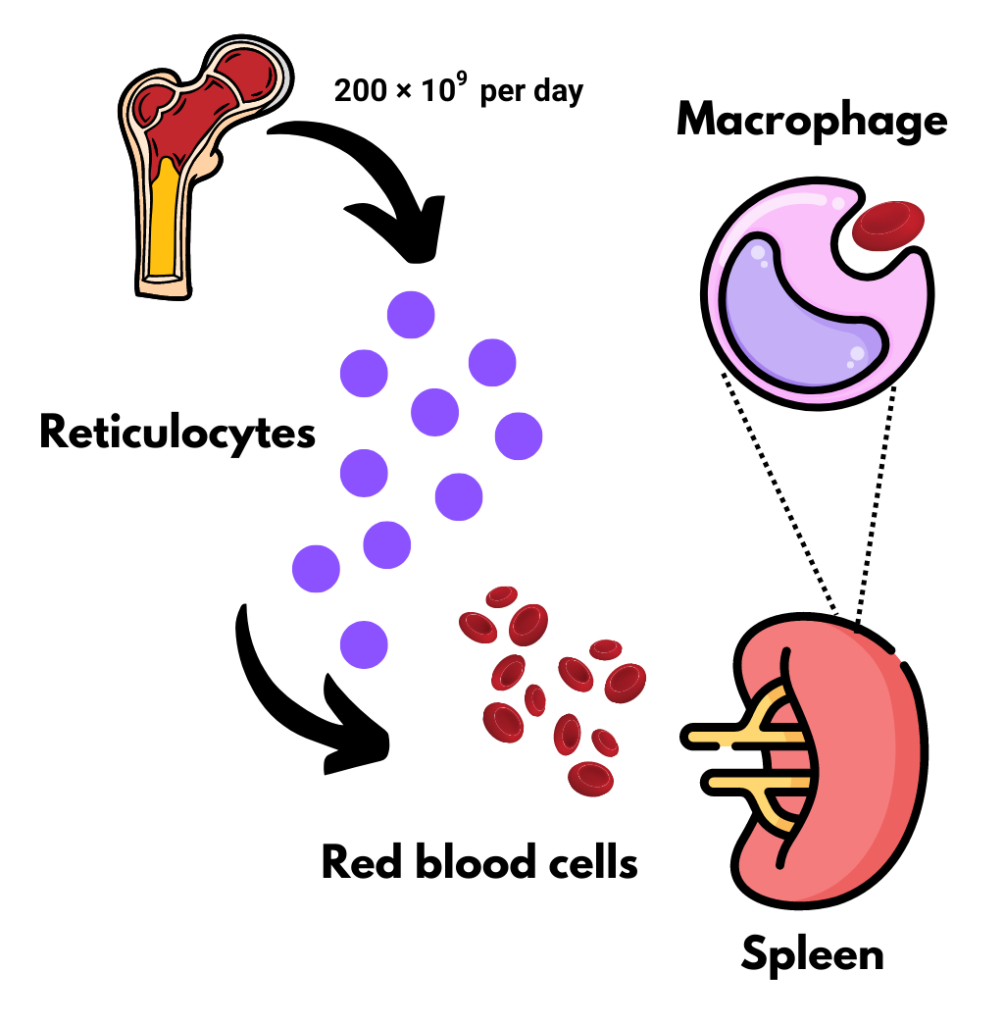
About 200 × 109 per day
There are 8 enzymes in the heme biosynthesis pathway. Mutations in each enzyme can cause porphyria. Therefore there are 8 different kinds of porphyria.
1-2 mg is derived from intestinal absorption.
Each unit of RBCs (processed from 420 mL of donor blood) contains about 200 mg of iron (0.47 mg iron/mL of whole donor blood or 1.08 mg iron/mL of pure RBCs).
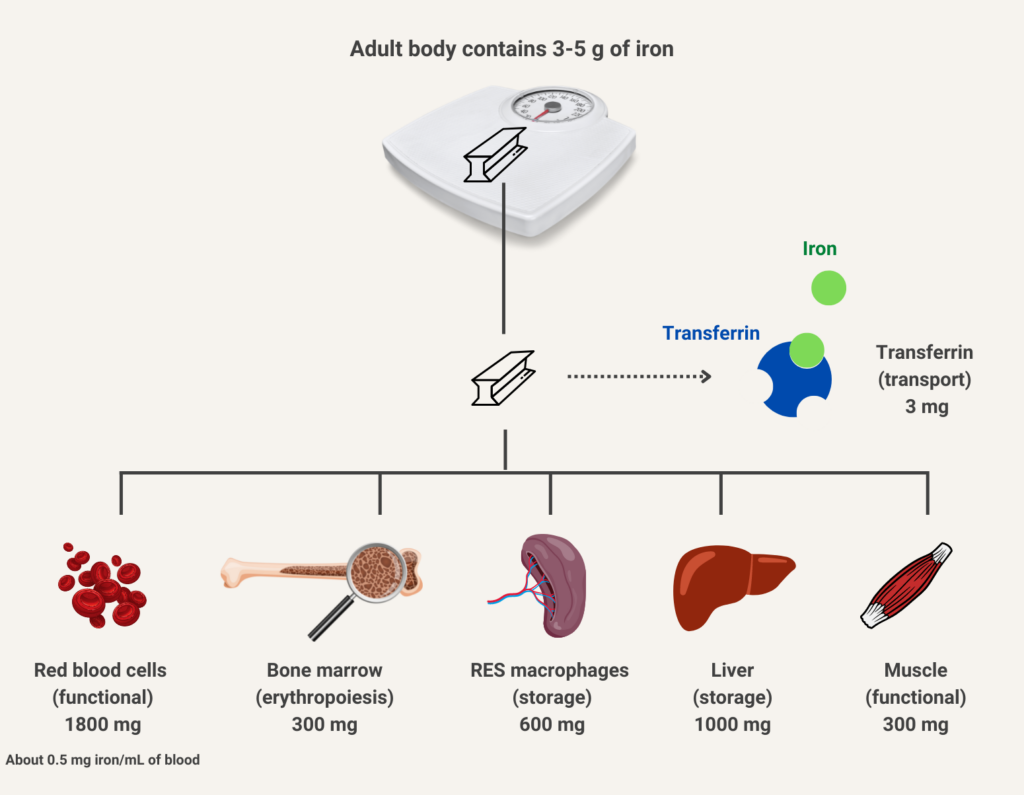
The human body normally contains 40 to 50 mg/kg of iron, which amounts to 3-4 grams. It is distributed in the following compartments:
- Hemoglobin in RBCs – about 30 mg/kg
- Myoglobin in muscle – about 4 mg/kg
- Iron-containing enzymes – about 2 mg/kg
- Storage in ferritin and hemosiderin – 0-2,000 mg
- Plasma transferrin – 1-2 mg
About 25 mg of iron daily (far more than the 1 mg that is absorbed by the gastrointestinal track).
25-40% larger; mean cell volume about 120 fL, though quite variable. Learn more here.
About 1%. This route of absorption is unaffected in patients with pernicious anemia, and is the rational for oral vitamin B12 therapy. Learn more here.
About 10%-20% of cases remain unexplained.
Hemoglobin typically increases by 20 g/L at 2 weeks, with a normal hemoglobin level usually achieved in 2 months. Ferritin may take up to 6 months to return to normal.
Historically, doses of elemental iron as high as 100–200 mg per day across two to three divided doses were recommended. However, it is now recognized that a dose of iron will increase hepcidin levels, which will then inhibit the absorption of the next dose. Iron absorption is most efficient with intermediate doses and on alternate days, and this approach is recommended in patients with mild symptoms, or no or mild anaemia.
Up-to-Date recommendations:
- We typically advise our patients to take their dose every other day as long as they can manage the schedule appropriately; a reasonable variation on the schedule that is easier to follow is to give the dose on Monday, Wednesday, and Friday.
- There is no reason to give more than one dose per day.
- The amount of iron in the every-other-day dose or the Monday-Wednesday-Friday dose is also not well established. However, there is not a reason to think higher doses improve absorption, and adverse effects are generally dose related. Thus, we typically use one tablet per dose (e.g. 325 mg ferrous sulfate).
2021 British Society of Gastroenterology guidelines for the management of iron deficiency anaemia in adults:

Learn more here.
IV carbohydrate loading has potential risk of hyponatremia, which can lead to cerebral edema.
According to the British Society of Haematology:
- Hct > 52% in men persisting for > 2 months
- Hct > 48% in women persisting for > 2 months
No, it must be ordered separately.
Women have a higher prevalence compared to men.
Learn more here.
Yes, and this is the justification for periodic surveillance endoscopies in these patients. Learn more here.
Yes. Vitamin B12 refers to a specific group of cobalt-containing corrinoids with biological activity in humans. This group of corrinoids is also referred to as cobalamins. The main cobalamins in humans and animals are adenosylCbl, methylCbl, and hydroxoCbl. Food Cbl is hydroxoCbl. Cyanocobalamin is a synthetic form of vitamin B12 found only in supplements. Learn more here.
Yes, typically for complement only. However, it is also weakly positive for IgG in about one quarter of cases. Learn more here.
X-linked. Learn more here.
No, about 30% of cases occur in patients without anemia. This is called isolated macrocytosis.
Oral vitamin B12 replacement at 1000 μg daily is an adequate alternative to IM B12 injections. Learn more here.
No, it is also seen in preadolescents and during pregnancy (more common at the beginning of pregnancy than in late pregnancy). Learn more here.
Yes, on both venous and arterial sides of the circulation. Learn more here.
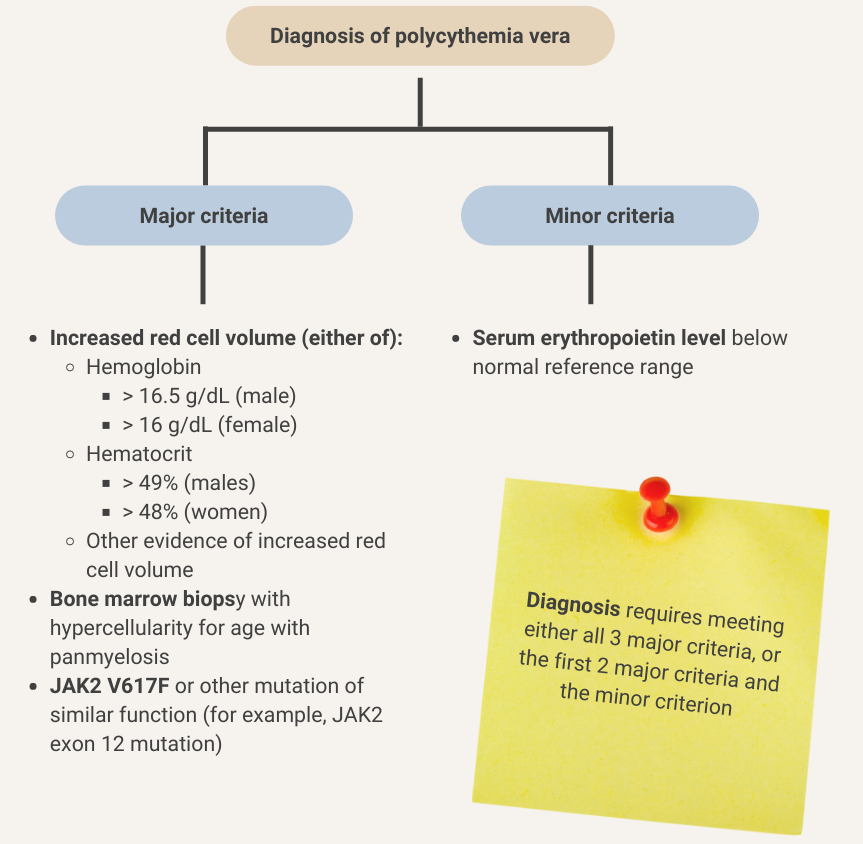
Serum erythropoietin is low in about 90% of patients with polycythemia vera. Learn more here.
Retinopathy has been reported in 70% of patients with HbSC compared with about 45% in sickle cell anemia. Learn more here.
No. It is also seen in:
- Pregnancy
- Chronic kidney failure
- Major depressive disorder
- Generalized anxiety disorder
- Panic disorder
- Attention-deficit/hyperactivity disorder
Read more here.
No. Serum iron levels are highly dependent on recent food intake and they follow a diurnal rhythm (though there is no evidence that fasting samples perform better than random samples). Learn more here.
There is no strong evidence for plasma therapy efficacy in atypical hemolytic uremic syndrome.
Learn more here.
No. Learn more here.
1-5 mg/day
According to DailyMed: Daily doses greater than 1 mg do not enhance the hematologic effect, and most of the excess is excreted unchanged in the urine. The usual therapeutic dosage in adults and children (regardless of age) is up to 1 mg daily. Resistant cases may require larger doses.
Guidance varies by professional organization.
2021 AGA clinical practice guidelines on the gastrointestinal evaluation of iron deficiency anemia:

2021 British Society of Gastroenterology (BSG) guideline on management of iron deficiency:

Hemoglobin (Hb) and hematocrit (Hct). Hct correlates a little better with red cell mass compared with Hb (learn more here).
Depends on the clinical context. In patients with anemia, we should refer to the Hb because oxygen carrying capacity is limiting, whereas the Hct should be considered in those with polycythemia since blood viscosity is limiting. For those with normal Hb/Hct, both oxygen carrying capacity and blood viscosity are at equipoise, so take your choice!
2021 AGA Clinical Practice Update on the Diagnosis and Management of Atrophic Gastritis (AG): Expert Review:
Providers should consider performing endoscopic surveillance every 3 years in patients with advanced AG. However, it should be recognized that optimal surveillance intervals remain to be determined, and shorter or longer intervals may be appropriate depending on individual risk assessment.

Learn more here.
2020 AGA Clinical Practice Guidelines on the Gastrointestinal Evaluation of Iron Deficiency Anemia:

2021 British Society of Gastroenterology guidelines for the management of iron deficiency anaemia in adults:

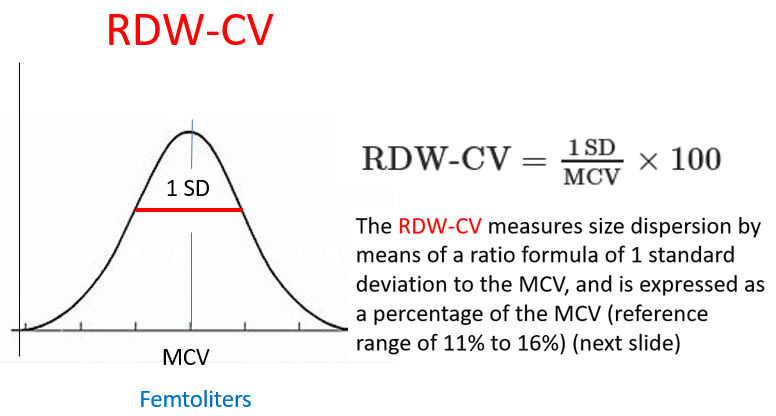
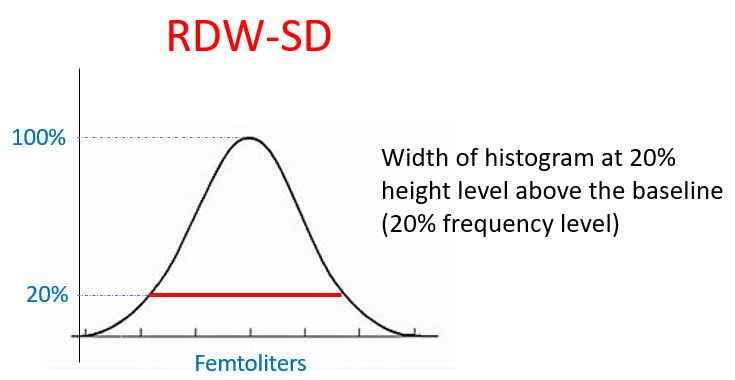
The RDW-CV (RDW coefficient of variation) is inversely proportional to the mean cell volume (MCV). As a result, patients with microcytosis have elevated RDW-CV regardless of variation in cell size, while those with macrocytosis have lower RDW-CD values. By contrast, the RDW-SD (RDW standard deviation) is not influenced by the MCV and therefore may be preferable to use. An RDW-SD > 46 fL represents anisocytosis.
Hydroxyurea (since 1998), L-glutamine, crizanlizumab, voxelotor
- Acute hemolytic transfusion reaction (AHTR)
- Febrile nonhemolytic transfusion reactions (FNHTR)
- Urticaria
- Anaphylaxis
- Transfusion-related acute lung injury (TRALI)
- Transfusion-associated circulatory overload (TACO)
- Nonimmune hemolysis
- Hypotensive transfusion reactions
- Transfusion-associated sepsis
Blood loss, hemolysis, erythropoietin administration, high altitude
- Hemorrhage +/- iron deficiency
- Hypersplenism
- Alcohol
- Bone marrow failure and aplastic anemia develop after an episode of hepatitis
- Complication of treatment of chronic hepatitis C
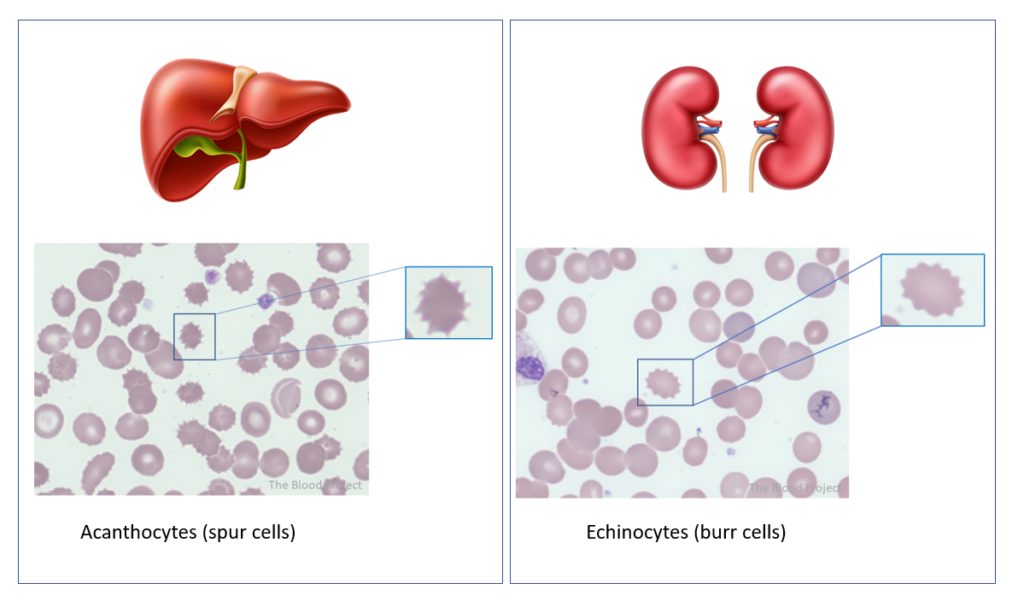
- Spur cell anemia
Learn more here.
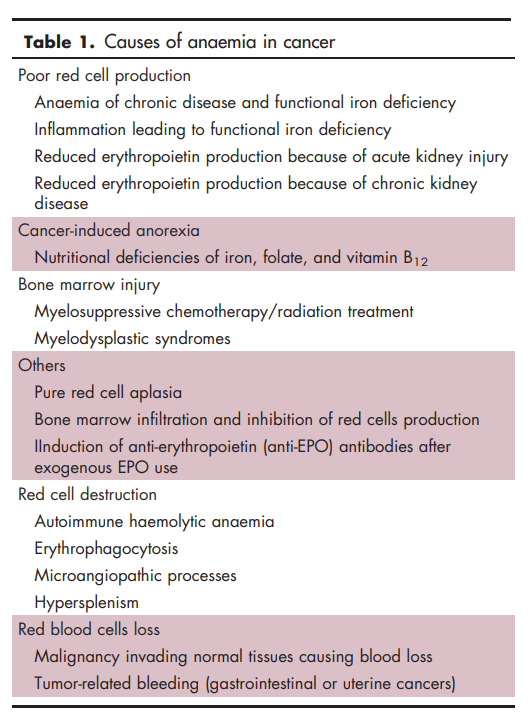
Learn more here.
- Changes in hemoglobin oxygen affinity, for example high-affinity hemoglobin mutations.
- Mutations in oxygen sensing/hypoxia-inducible factor (HIF) signaling pathway, for example Chuvash polycythemia (mutation in VHL gene, automosomal recessive).
Learn more here
Primary
- Congenital (erythropoietin receptor mutations)
- Polycythemia vera
- Idiopathic erythrocytosis
Secondary
- Congenital
- Defects in oxygen sensing pathway – Chuvash erythrocytosis (VHL mutation)
- Left shift of hemoglobin (Hb) oxygen dissociation curve:
- High affinity Hb
- 2,3-DPG deficiency
- Acquired
- Hypoxia-driven:
- Central process:
- Chronic lung disease
- Right-to-left cardiopulmonary shunts
- Carbon monoxide poisoning
- Smoking
- Sleep apnea
- High altitude
- Local process:
- Renal artery stenosis
- Hydronephrosis
- Renal cysts
- Central process:
- Pathological erythropoietin production:
- Tumors:
- Hepatocellular carcinoma
- Renal cell cancer
- Cerebellar hemangioblastoma
- Uterine leiomyoma
- Pheochromocytoma
- Meningioma
- Tumors:
- Drug-associated:
- Erythropoietin
- Androgens
- Diuretics
- Hypoxia-driven:
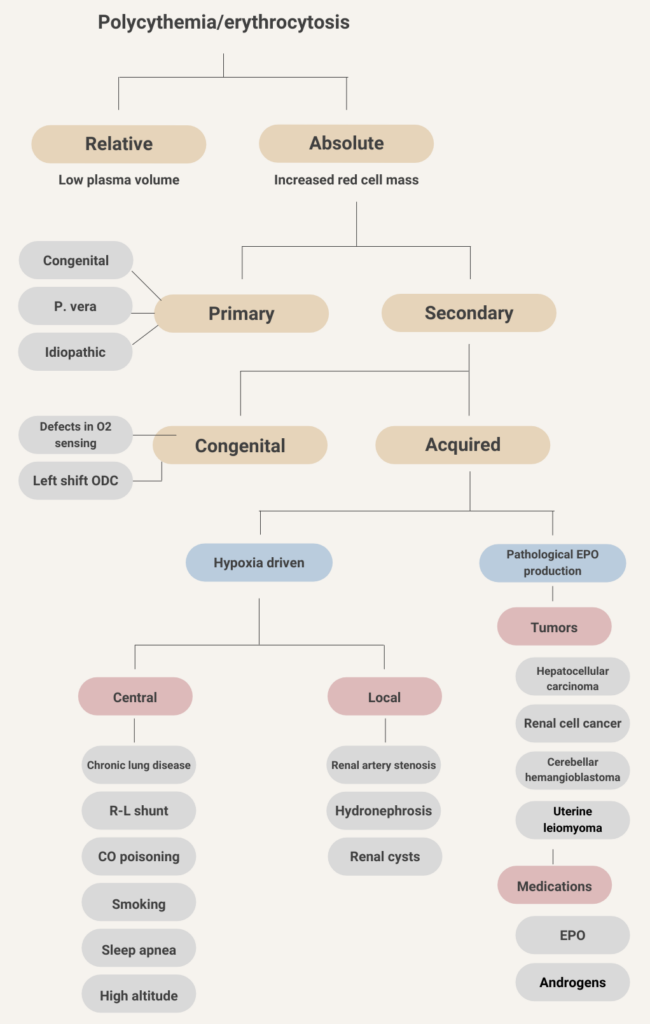
Learn more here.
Nucleated red blood cells are a reflection of extreme increases in erythropoietic activity as seen in/with:
- Hemoglobinopathies
- Brisk hemolysis
- Rapid blood loss
- Other conditions of hematopoietic stress such as sepsis
- Damage or stress to bone marrow, for example in:
- Chronic myeloid leukemia
- Acute leukemia
- Myelodysplastic syndromes
- Chemotherapy
- Myelophthisic conditions, including:
- Metastatic cancer to bone marrow
- Bone marrow fibrosis

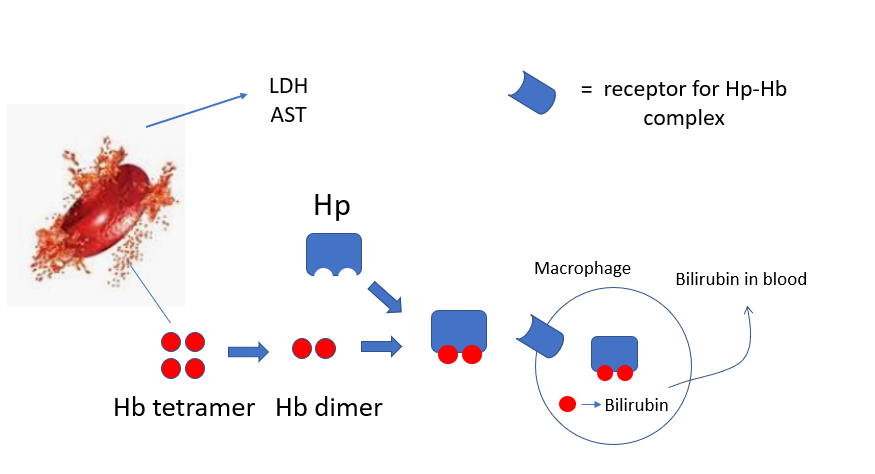
Lactate dehydrogenase (LDH), indirect (unconjugated) bilirubin, haptoglobin, and AST. Learn more here.
- Nausea
- Vomiting
- Diarrhea
- Constipation
- Epigastric pain
- Metallic taste
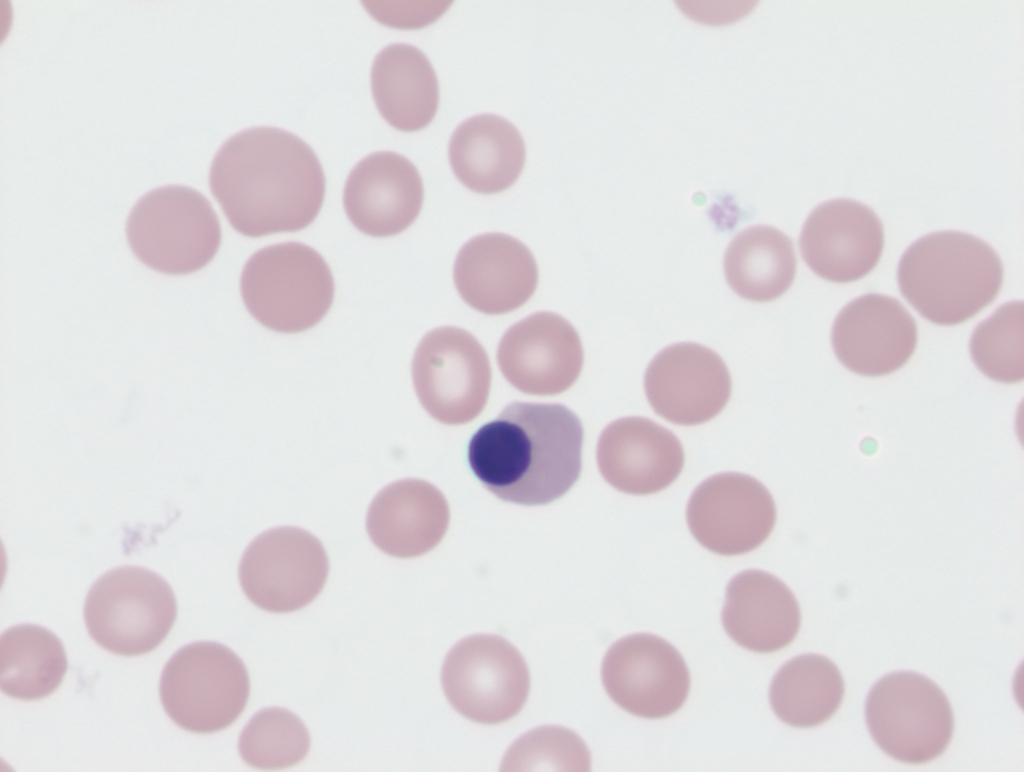
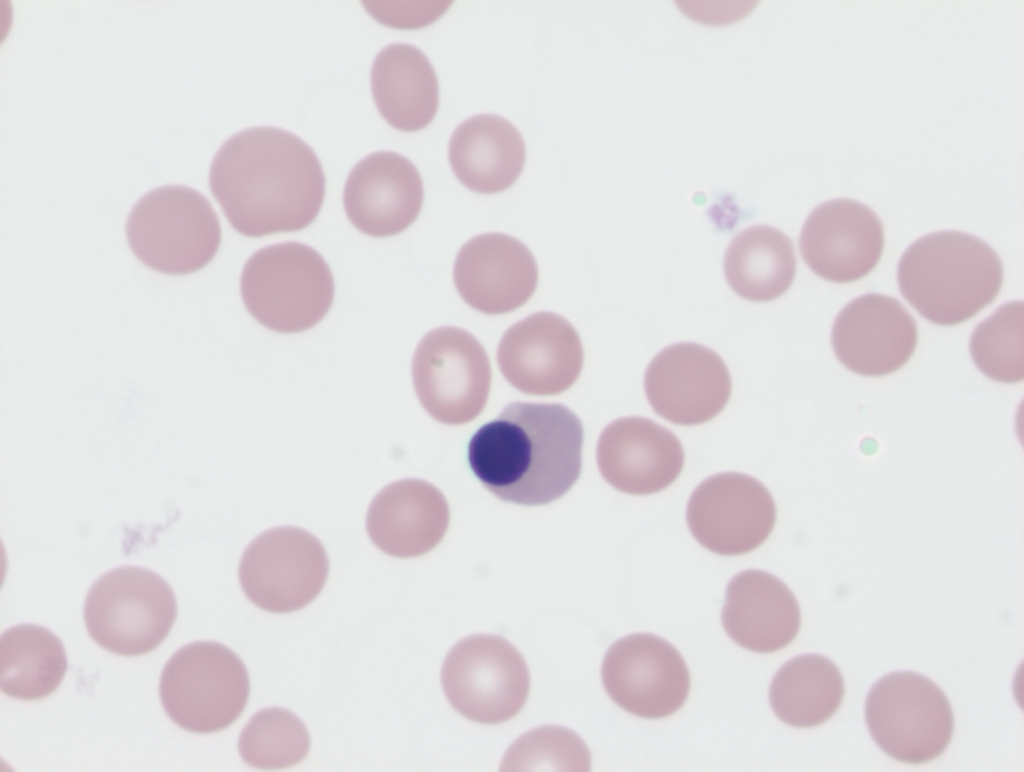
Large precipitates of denatured hemoglobin often seen in patients with G6PD deficiency and hemolysis. Requires special stain of blood using methyl violet. See images.
- Those at risk for transfusion-associated graft-versus-host disease (TA-GVHD) caused by proliferation of donor T lymphocytes
- Those who are immunocompromised
- Those receiving intrauterine transfusion
- Those with hematologic malignancies or solid tumors, including:
- Sarcoma
- Neuroblastoma
- Hodgkin lymphoma
- Those who are recipients of marrow or peripheral blood stem cell transplantation
- Those receiving RBCs from blood relatives or human leukocyte antigen-compatible donors
- Those receiving fludarabine therapy
- Those receiving granulocyte transfusions
According to the AABB:
Description:
- Blood components that contain viable lymphocytes may be irradiated to prevent proliferation of T lymphocytes, which is the immediate cause of TA-GVHD.
- Irradiated blood is prepared by exposing the component to a radiation source.
- The standard dose of gamma or X-ray irradiation is 2500 centigray (cGy) targeted to the central portion of the container with a minimum dose of 1500 cGy delivered to any part of the component.
Indications:
- Patients at risk for TAGVHD, including:
- Fetal and neonatal recipients of intrauterine transfusions
- Selected immunocompromised recipients
- Recipients of cellular components known to be from a blood relative
- Recipients who have undergone peripheral blood progenitor cell transplantation
- Recipients of cellular components whose donor is selected for HLA compatibility and recipients of granulocyte transfusions.
- Patients receiving purine analogues (eg, fludarabine, cladribine) or certain other biological immunomodulators (eg, alemtuzumab, antithymocyte globulin) who may be at risk for TA-GVHD, depending on clinical factors and the source of the biological agent.
- Symptomatic hemolytic anemia
- Presence of gallstones
- Large reduction in exercise tolerance
- Growth retardation
- Skeletal changes or leg ulcers due to HS
- Extramedullary hematopoietic tumors
- Vascular compromise of vital organs in older patients
- Splenic infarct with pain or early satiety from splenomegaly
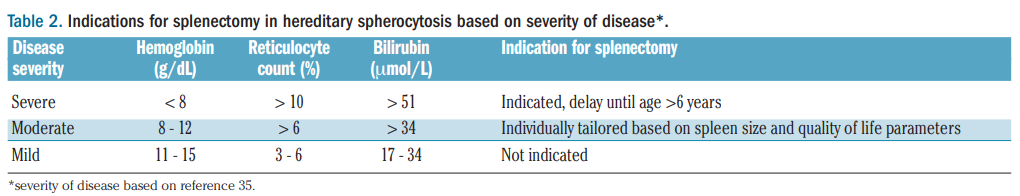
Consider delaying splenectomy until > 6 years old if possible.
Guideline recommendations:
Recommendations regarding splenectomy in hereditary hemolytic anemias by Splenectomy in Rare Anemias Study Group (2017):


2012 British Committee Standards in Haematology (BCSH) expert guideline on diagnosis of hereditary spherocytosis:
- Patients intolerant or not responding to oral iron.
- When there is a need for a quick recovery in patients with iron deficiency anemia.
- Patients taking erythropoiesis stimulating agent (ESA), for example those with anemia of chronic kidney disease.
- Patients with inflammatory bowel disease inflammatory bowel disease and iron deficiency anemia.
- Patients with history of severe allergic reactions to plasma-containing products.
- Patients who have absolute immunoglobulin A (IgA) deficiency and for whom no IgA-deficient RBCs are available.
- Patients at risk of hyperkalemia.
- Neonates with neonatal alloimmune thrombocytopenia requiring maternal RBC transfusion that contains antihuman platelet antigen-1a (however, use of washed RBCs is not required.)
According to AABB:
- Description:
- Washed components are typically prepared using 0.9% Sodium Chloride, Injection USP with or without small amounts of dextrose.
- Washing removes unwanted plasma proteins, including antibodies and glycerol from previously frozen units.
- The shelf life of washed components is no more than 24 hours at 1 to 6 C or 4 hours at 20 to 24 C.
- Washing is not a substitute for leukocyte reduction, and only cellular components should be washed.
- Indications:
- To reduce exposure to antibodies targeting known recipient antigens.
- To remove constituents that predispose patients to significant or repeated transfusion reactions (eg, removal of IgA-containing plasma in providing transfusion support for an IgA-deficient recipient or in rare recipients experiencing anaphylactoid/anaphylactic reactions to other plasma components).
Less avascular necrosis, pulmonary hypertension, leg ulcers, and stroke; more retinopathy. 0.4% painful crises per patient year in those with HbSC, less than half the rate in sickle cell anemia. Learn more here.
| Complication | HbSC | HbSS |
|---|---|---|
| Avascular necrosis | + | +++ |
| Pulmonary hypertension | + | +++ |
| Leg ulcers | + | +++ |
| Stroke | + | +++ |
| Retinopathy | +++ | + |
| Painful crises | + | +++ |
Conditions associated with:
- Appropriate release of erythropoietin from oxygen sensing cells in the kidney, including:
- Cardiopulmonary disease
- High altitude
- Hereditary hemoglobin mutations associated with high oxygen affinity
- Inappropriate release of erythropoietin from:
- Oxygen sensing cells in the kidney, including renal disorders and drugs
- Tumor cells
See additional list of causes from PMC7829024. Learn more here.
- Neurological symptoms
- Pancreato-intestinal involvement
- Gangrene of the fingers or toes
- Ulcerative–necrotic skin lesions
- Myocardial infarction
- Ischemic cardiomyopathy
Learn more here.
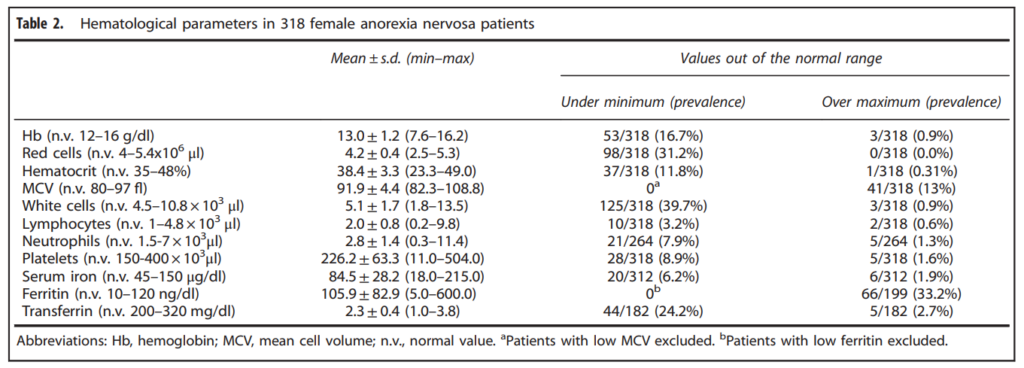
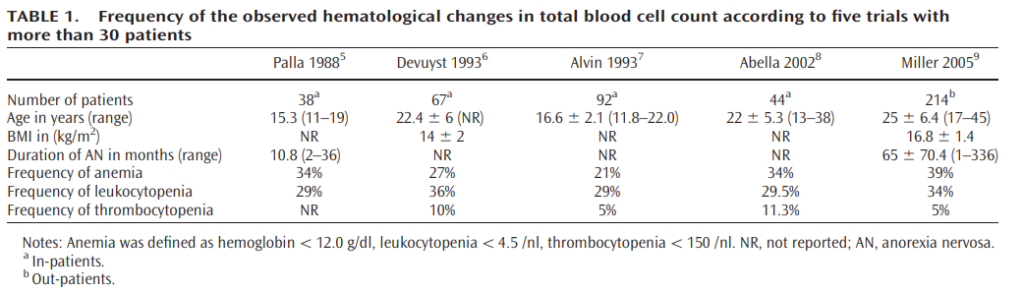
- Anemia (21%-39% of patients)
- Typically normocytic, normochromic)
- Rarely acquired hemolytic syndrome from hypophosphatemia accompanying anorexia nervosa (e.g., during the refeeding process)
- Leukopenia (29%-36% of patients)
- Mild neutropenia
- Lymphocytopenia
- Thrombocytopenia (5-11% of patients)
- Acanthocytes on peripheral smear
- Gelatinous transformation or serous atrophy on bone marrow exam
Learn more here.
- Increase in cardiac output, primarily from increased stroke volume
- Increase in 2,3-diphosphoglycerate (2,3-DPG), leading to shift of oxygen dissociation curve to the right (lower affinity for oxygen, increased oxygen unloading in tissues)
- Increase in oxygen extraction by tissues
- Redistribution of blood flow to vital tissues such as the brain and heart

- Renal artery stenosis
- Hydronephrosis
- Renal cysts
- Renal tumors
- Cerebellar hemangioblastoma
- Hepatocellular carcinoma
- Uterine leiomyoma
- Renal cell carcinoma
- Meningioma
- Fatigue
- Pallor
- Dizziness
- Headaches
- Vertigo
- Tinnitus
- Dyspnea
- Inactivity
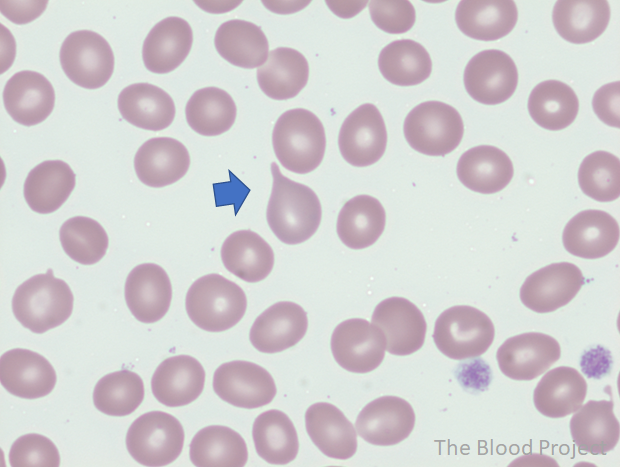
Teardrop cells are red cells appearing in the shape of a teardrop or a pear with a single, short or long, often blunted or rounded end are called teardrop cells. True tear drops have blunted tips and point in different directions. Teardrop cells are commonly seen in chronic idiopathic myelofibrosis.
Hemoglobinuria and myoglobinuria
Type I cells – red blood cells (RBCs) express GPI-APs (for example, CD59) at normal density
- RBCs express normal amounts of GPI-anchored proteins (GPI-AP), such as CD59
- Full protection against complement-mediated lysis
- Normal RBC lifespan of about 120 days
Type II cells – red cells partly deficient in GPI-APs;
- RBCs partly deficient in GPI-APs
- Partial protection against complement-mediated lysis
- Lifespan intermediate between type1 and type 3 cells
Type III cells – red cells completely deficient in GPI-APs.
- RBCs completely deficient in GPI-APs
- No protection against complement-mediated lysis
- RBC lifespan 10-15 days
- Acute intermittent porphyria (AIP)
- Hereditary coproporphyria (HCP)
- Variegate porphyria (VP)
- ALA-dehydratase deficiency porphyria (ADP)
- Reduce rates of alloimmunization
- Reduce rates of febrile nonhemolytic transfusion reactions
- Prevent cytomegalovirus transmission
Another perspective:
- Mean cell volume (MCV)
- Mean corpuscular hemoglobin (MCH)
- Mean corpuscular hemoglobin concentration (MCHC)
- Red cell distribution width (RDW)
Learn more here.
What are the goals of treatment in a patient with acute porphyria presenting with an acute attack?
- Eliminate precipitating factors
- Treat patient’s symptoms
- Reduce ALAS-1 activity and production of 5-aminolevulinic acid (ALA) and porphobilinogen (PBG) using:
- Carbohydrate loading
- Heme infusions
Thrombosis and disease progression to myelofibrosis or acute leukemia. Learn more here.
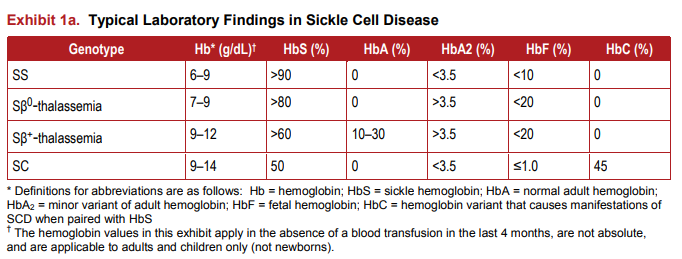
- Homozygous sickle cell disease (HbSS)
- Sickle beta0 thalassemia
- Sickle hemoglobin C disease
- Sickle beta+ thalassemia
- Thrombophlebitis
- Coagulopathy
- Hepatic iron overload (with chronic use)
Learn more here.
Acquired mutation in the X-linked phosphatidylinositol glycan class A (PIGA) gene in an hematopoietic stem cell, leading to deficiency of GPI-anchored proteins on hematopoietic cells, including CD55 and CD59, which in turn leads to activation of the alternative complement pathway and complement-mediated hemolysis. Learn more here.
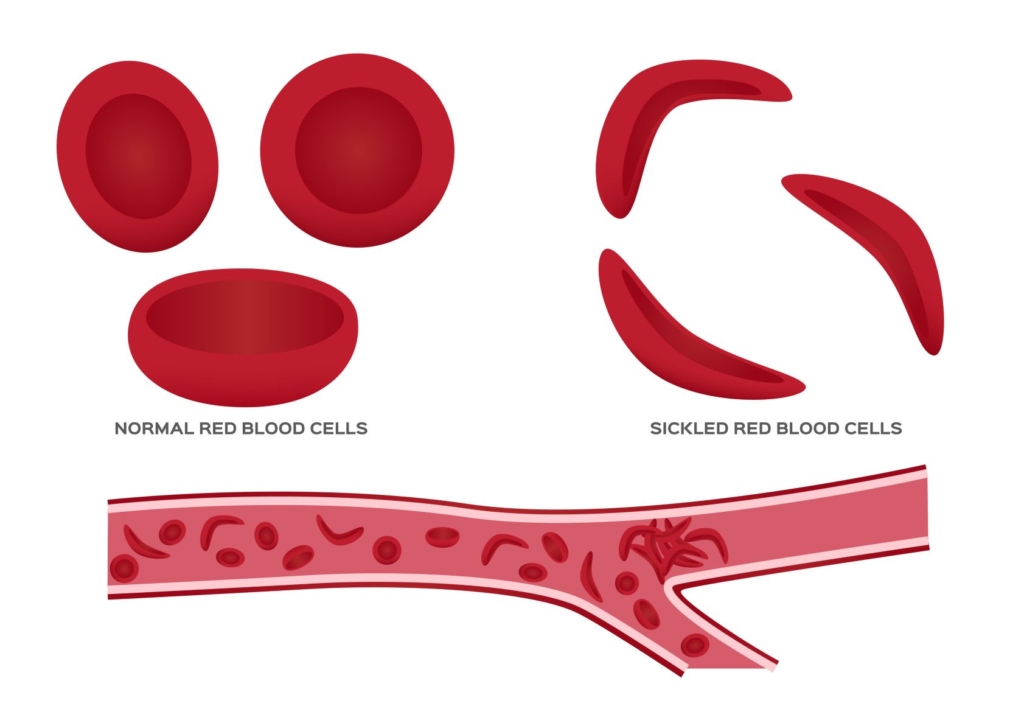
Aggregation of sickled cells, causing vaso-occlusion of small blood vessels, leading, in turn, to ischemia-reperfusion injury.

- Conditions involving macrophage-mediated ingestion or red cells:
- Hemophagocytic lymphohistiocytosis (HLH)
- Pernicious anemia
- Resorbing hematoma
- Conditions not associated with red cell destruction:
- Cirrhosis may also mimic hemolysis because the liver is responsible for synthesizing haptoglobin and hepatocytes release LDH, and AST may be elevated disproportionately to ALT, especially in alcoholic liver disease.
- Iron levels
- Iron increases hepcidin levels.
- Represents a feedback mechanism to maintain stable body iron levels.
- Inflammation
- Leads to increased hepcidin levels.
- Represents a host defense mechanism to limit extracellular iron availability to microbes.
- Erythropoiesis
- Increased erythroid activity leads to decreased hepcidin levels.
- Ensures iron supply for erythropoiesis.
- Erythropoietic drive overrides both iron sensing and inflammation sensing mechanisms to control hepcidin levels.
Autoimmune hemolytic anemia
Acute intermittent porphyria
Direct antiglobulin test
Erythropoietin
Glucose-6-phosphate dehydrogenase. Learn more here.
Microangiopathic hemolytic anemia
Paroxysmal nocturnal hemoglobinuria
Packed red blood cells, used for transfusion.
Polycythemia vera
Red cell distribution width
Sickle cell anemia
Sickle cell disease
- Hypochromic microcytic red cells
- Pencil cells, which is a type of elliptocyte, reported in about two-thirds of cases
- Target cells
- Fragments (in severe cases)
- Thrombocytosis (in about 10% of cases)
Transferrin saturation. Represents the % of iron binding sites on serum transferrin that are bound by iron atoms.
Vaso-occlusive crisis, occurs in patients with sickle cell disease (SCD). The most frequent cause of recurrent morbidity and SCD-related admission to hospital.
Measured:
- Hemoglobin (Hb)
- Red blood cell (RBC) count
- Mean cell volume (MCV)
Calculated:
- Hematocrit (Hct) = MCV x RBC count
- Mean corpuscular hemoglobin (MCH) = Hb/RBC count
- Mean corpuscular hemoglobin concentration (MCHC) = Hb/Hct
- Hormonal fluctuations during the menstrual cycle
- Fasting (reduction in calorie intake)
- Smoking
- Infections
- Alcohol
- Exposure to porphyrinogenic drugs including:
- Antibiotics
- Oral contraception
- Anticonvulsants
Read more here.
In one study:
- Normal platelet count in 84.6%
- Thrombocytosis (> 400 × 10 9/L) in 13.3%
- Thrombocytopenia (< 150 × 10 9/L) in 2.1%
They become smaller, they acquire a larger surface-to-volume ratio, they lose their organelles and the mean corpuscular hemoglobin concentration increases.

British Society for Haematology recommends investigating patients with persistently elevated venous hematocrit (Hct) (> 52% in males and > 48% in females)
FDA test requirements (with American Red Cross guidance):
- Hepatitis B virus (HBV):
- The tests used for blood donor screening are the GS HBsAg EIA 3.0, a qualitative ELISA for the detection of Hepatitis B Surface Antigen (HBsAg), and the Ortho HBc ELISA for the qualitative detection of antibody to HBV core antigen (anti-HBc) in human serum and plasma samples.
- An FDA licensed triplex nucleic acid test (NAT) using transcription-mediated amplification was introduced by the Red Cross in 2009. The assay detects HBV DNA, HIV-1 RNA, and HCV RNA.
- The frequency of detecting an active HBV infection in a blood donor is about 1 per 12,000 donations screened.
- The per-unit risk of HBV infection through blood transfusion is less than 1 per million units screened.
- Hepatitis C (HCV):
- The test used for blood donor screening is the Ortho HCV ELISA for the qualitative detection of antibody to HCV antibodies (anti-HCV) in human serum or plasma samples.
- A duplex nucleic acid test (NAT) was introduced for HIV-1/HCV RNA detection in 1999 and updated to include the detection of HBV DNA in 2009.
- Donors who test HCV-antibody reactive, but NAT nonreactive by routine testing are further tested individually for HCV RNA by NAT.
- Donors who test anti-HCV and HCV NAT reactive do not require further testing.
- The frequency of detecting a positive donor is about 1 per 5,000 donations screened.
- The per-unit risk of HCV infection through blood transfusion is less than 1 per 2 million units screened.
- HIV:
- The test used for blood donor screening is the GS HIV-1/HIV-2 PLUS O EIA for the simultaneous qualitative detection of anti-HIV 1 (groups M and O) and/or HIV-2 in human serum or plasma.
- A duplex nucleic acid test (NAT) was introduced for HIV-1/HCV RNA detection in 1999 and updated to include the detection of HBV DNA in 2009.
- Donors who test antibody reactive are further evaluated by additional tests to confirm the presence of HIV antibody and to differentiate HIV-1 from HIV-2 antibodies.
- Donors who test anti-HIV-1/HIV-2 and HIV-1 NAT reactive are not further tested.
- The frequency of detecting HIV-1 in a blood donor is about 1 per 33,000 donations screened.
- detecting HIV-2 in a blood donor is extremely rare at 1 per 57 million donations, with only 5 such infected donors ever identified since HIV-2 screening began in 1992.
- The per-unit risk of HIV-1 infection through blood transfusion is less than 1 per 2 million units screened.
- Human T-cell lymphotropic virus I/II (HTLV-I/II):
- The test used for blood donor screening is the Avioq HTLV-1/2 Microelisa system for the qualitative detection of antibodies to HTLV-1 and HTLV-2 in human serum or plasma samples.
- Donors who test reactive for anti-HTLV-1/2 are further tested using an FDA licensed western blot to determine if antibodies are present.
- There are no nucleic acid tests (NAT) available for HTLV-1/2.
- The per-unit risk of transfusion-transmitted HTLV-1/2 is less than 1 per 2 million units screened, and the frequency of detecting an infected donor is 1 per 27,000 donations screened.
- Syphilis:
- Screening for syphilis is performed using a qualitative test that detects the presence of antibodies to the spirochete (corkscrew-shaped bacterium), Treponema pallidum, by an automated agglutination assay based on specific pattern recognition. C
- Confirmation is performed using another serologic test for total antibodies, an enzyme-linked immunoassay, as well as a test for reagin (a protein-like substance that is present during acute infection and for several months following resolution of infection).
- No cases of transfusion-transmitted syphilis have been recorded in more than 50 years.
- West Nile virus (WNV):
- WNV RNA is detected by an FDA licensed NAT assay similar to that used for HIV-1, HCV, HBV, and Zika virus.
- NAT-reactive donations are further tested by repeat NAT and antibody to confirm infection.
- Following the introduction of blood donor screening, there have been 15 cases of transfusion transmission from screened blood; all are believed to be due to donations having very low levels of virus. This translates to a risk of about 1 per 84 million donations for the Red Cross overall (or 1 per 35 million during the summer transmission season).
- Trypanosoma cruzi:
- The Red Cross blood donations are screened using the Ortho T. cruzi Enzyme-Linked Immunosorbent Assay (ELISA) for the qualitative detection of antibodies to T. cruzi in human serum or plasma samples.
- An FDA licensed enzyme strip immunoassay (ESA) is used for confirmatory testing.
- Because T. cruzi is not endemic in the United States, the Red Cross (and all US blood centers) donors are tested only once.
- The frequency of detecting a positive donor is about 1 per 15,000 first-time donations screened.
- Babesiosis:
- In May 2018, the Red Cross began testing from whole blood samples using a NAT assay that detects the main four species of babesia pathogenic to humans. The assay, now FDA licensed, detects ribosomal RNA of the parasite following red cell lysis, significantly increasing sensitivity and obviating the need for antibody testing.
- Tested in 14 states in the US where incidence of infection is highest.
Refers to changes in quality and quantity of packed red blood cells (pRBCs) during storage, including:
- Change in cell shape change and microvesiculation
- Decrease in adenosine triphosphate (ATP) and 2,3-diphosphoglycerate levels
- Increase in lysophospholipids
- Increase in potassium
- Increase in free hemoglobin (Hb)
The storage lesion represents a risk to efficient RBC perfusion and tissue oxygen delivery, and it has been suggested that it may result in adverse outcomes. This has led to the classification of “young” (<14-21 days) vs “old” (>21 days) RBC units. According to the AABB, 13 randomized control trials have evaluated the effect of RBC storage duration of transfused RBCs on patient outcomes (7 since 2012). However, there is currently no formal guidance on the optimal length of RBC storage prior to transfusion. The AABB recommends administering RBC units within licensed expiration date (standard issue) rather than transfusing only fresh RBCs (< 10 days old) to patients, including neonates.
Burr cells are red blood cells (RBCs) with 10-30 uniform, short, blunt projections distributed evenly around the circumference of the cell, giving the red cell surface a serrated appearance. The red blood cells retain central pallor. They are found in uremia, and to a lesser extent post splenectomy. Cells resembling burr cells (called crenated RBCs) occur when blood is stored too long in EDTA-containing tubes.

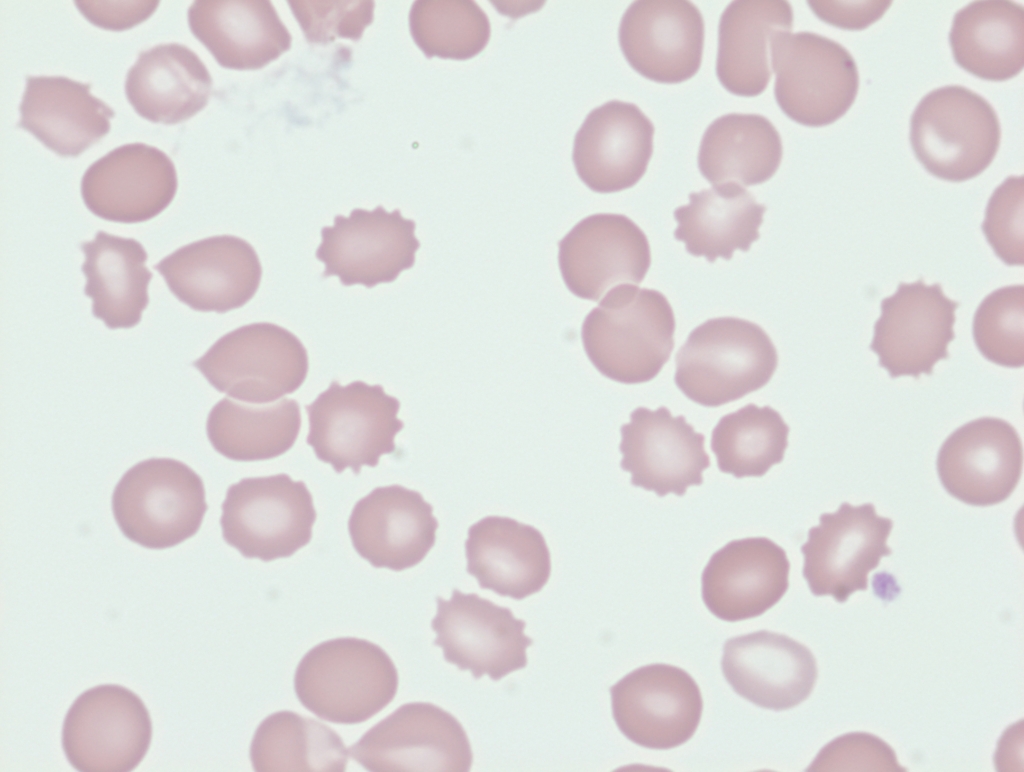
A cold reactive antibody that causes agglutination without antiglobulin antisera at 4 degrees C (39.2 degrees F).
The first automated counters to hit the market was the Coulter counter, named after its inventor, Wallace H. Coulter, an electrical engineer. These early counters used what is now known as the Coulter principle, which states that particles (for example a red blood cell) that pass through an orifice with an electric current, produce a change in impedance that is proportional to the volume of the particle. That change is registered and recorded. Coulter patented his method in 1953.

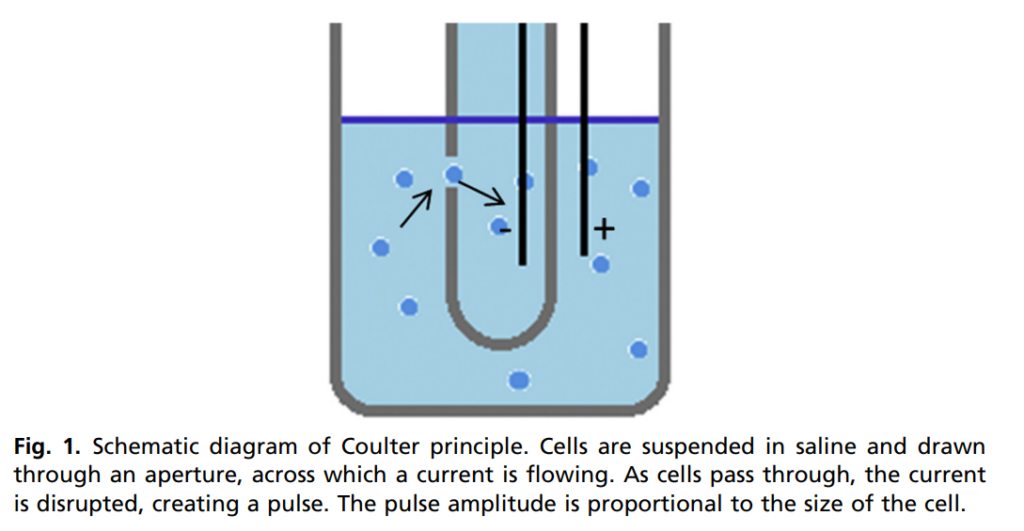
Serological or electronic crossmatches are performed to detect serological incompatibility (the presence of antibodies) between recipient and donor before transfusion.

According to the AABB:
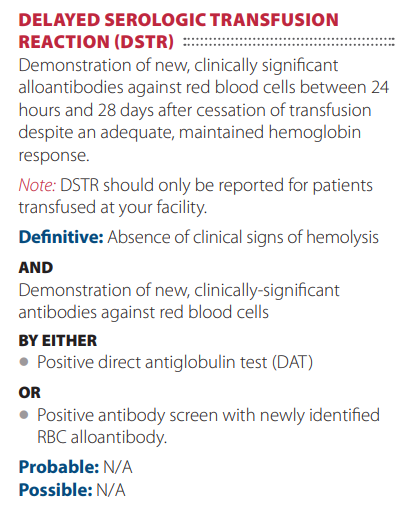
Defined as ≥ 1 degrees C increase in temperature ≥ 38 degrees C (100.4 degrees F) that is associated with transfusion and no other causes are evident. Caused by antibody to donor leukocytes. Symptoms include:
- Fever
- Chills
- Tachypnea
- Headache
- Vomiting
According to the AABB, febrile nonhemolytic transfusion reaction defined as fever and/or chills without hemolysis occurring
in the patient during or within 4 hours of cessation of transfusion. If transfusion-related, the most common cause is a reaction to passively transfused cytokines or a reaction of recipient antibodies and leukocytes in the blood product. If blood culture of patient or residual component is performed, the results should be negative. Laboratory findings should show no evidence of
acute hemolysis.

Treatment includes antipyretic such as acetaminophen or meperidine (for more severe reactions).
Administer leukocyte-reduced blood or washed RBCs to reduce risk.
Howell-Jolly bodies are nuclear remnants in red blood cells, typically about 1 μm in diameter. As such, they are composed of DNA. Their presence indicates asplenia. Learn more about the history of discovery of the Howell-Jolly body here.
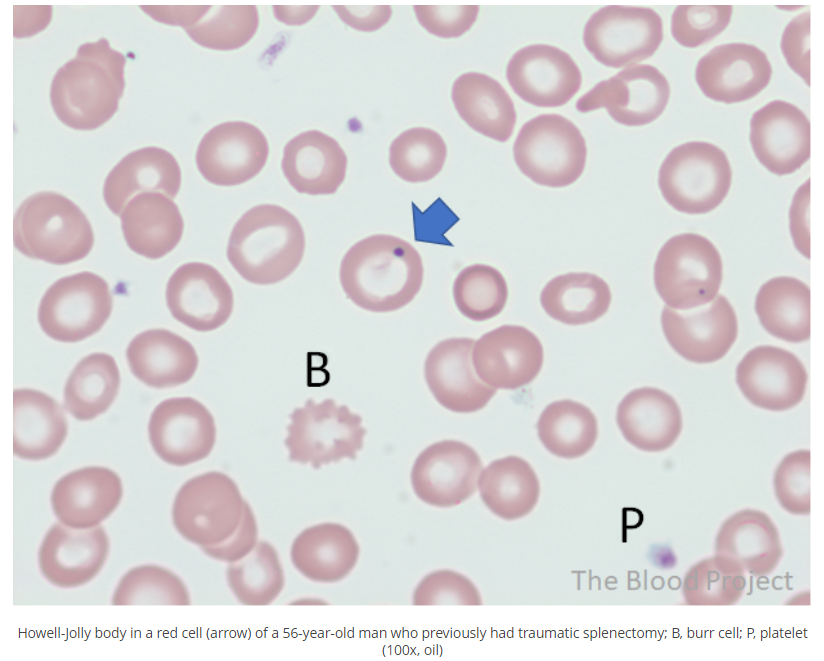
41% – 50% in male adults and 36% to 44% in female adults.
4.5-5.9 × 1012/L for male persons and 4.0-5.2 × 1012/L in female persons.
Note: 1012 = trillion!
On average about 1/3 of iron binding sites are occupied with iron atoms. Normal values, however, can range between 20-45%.
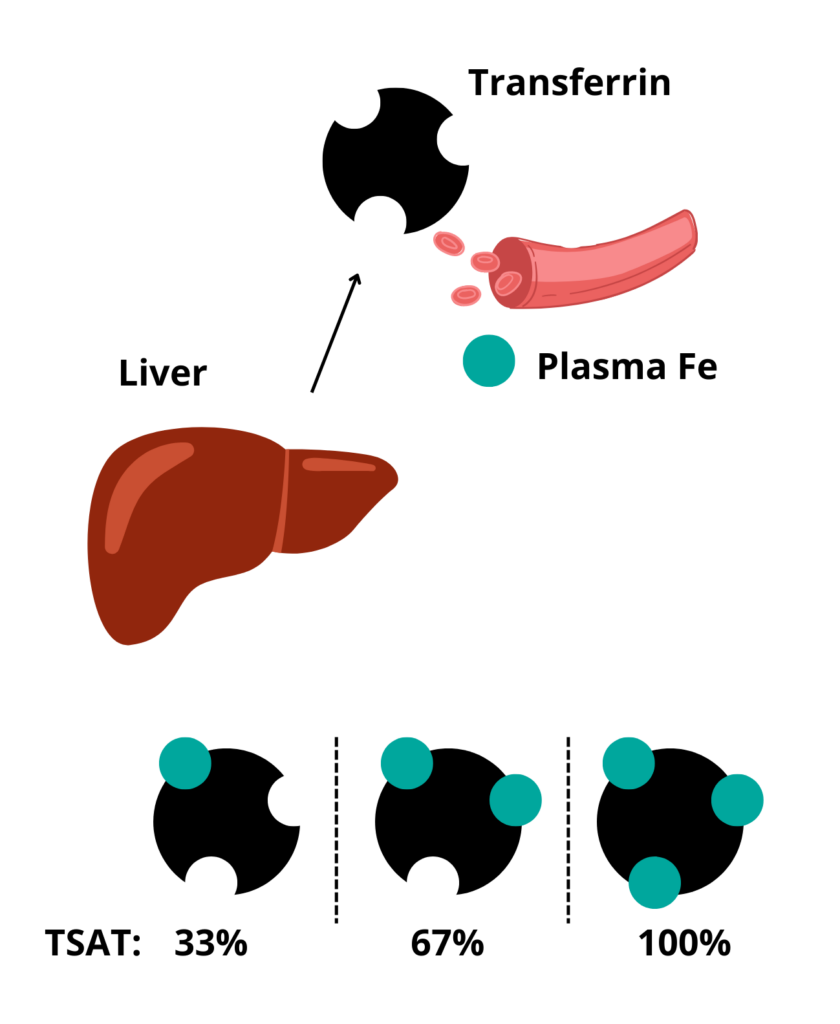
An abnormally shaped red cell
Examples of poikilocytes include:
- Sickle cells (drepanocytes)
- Burr cells (echinocytes)
- Spur cells (acanthocytes)
- Target cells (codocytes)
- Spherocytes
- Elliptocytes (includes pencil cells)
- Stomatocytes
- Bite cells
- Tear drop cells (dacrocytes)
- Schistocytes (includes helmet cells, horn cells, triangular cells, microspherocytes)
- Fish-shaped red cells
In patients with intense erythropoietic stimulation, “young” basophilic reticulocytes are released prematurely from the bone marrow into the peripheral blood, causing shortened reticulocyte maturation time in the bone marrow (sometimes to < 1 day), but a longer reticulocyte maturation time in the peripheral blood. The RPI is a corrected reticulocyte count that accounts for reticulocytes in all development stages.
Reticulocyte production (maturation) index = reticulocyte count [%] × patient’s packed cell volume/0.45) / maturation time in peripheral blood.
Maturation time (days) varies according to Hct:
| Hematocrit (%) | Maturation correction |
|---|---|
| < 15 | 2.5 |
| 16-25 | 2 |
| 26-35 | 1.5 |
| 36-45 | 1 |
Reticulocyte production index of > 3 suggests normal bone marrow response to anemia while < 2 suggests inadequate response.
See calculator.
Immature, nonnucleated red blood cell (RBC) that circulates in the blood before losing its RNA and differentiating into mature RBC (the maturation process in the circulation takes 1-3 days). Reticulocytes are larger than mature RBCs, and contain intracellular organelles (with the exception of a nucleus).
Schistocytes, or schizocytes (from the Greek word schisto, broken or cleft) are circulating fragments of red blood cells or red blood cells from which cytoplasmic fragments have been lost. They lack central pallor and are often deeply staining. For more information see. Additional images shown here.
IV transfusion without removing patient blood volume.
A target cell is a red cell with centrally located disk of hemoglobin surrounded by an area of pallor with an outer rim of hemoglobin adjacent to the cell membrane giving the cell the appearance of a bull’s eye or shooting target. Seen in liver disease (macrocytic targets), iron deficiency anemia, and thalassemia (microcytic target cells). Target cells may also be seen in hemoglobin C and E disease, and following splenectomy.
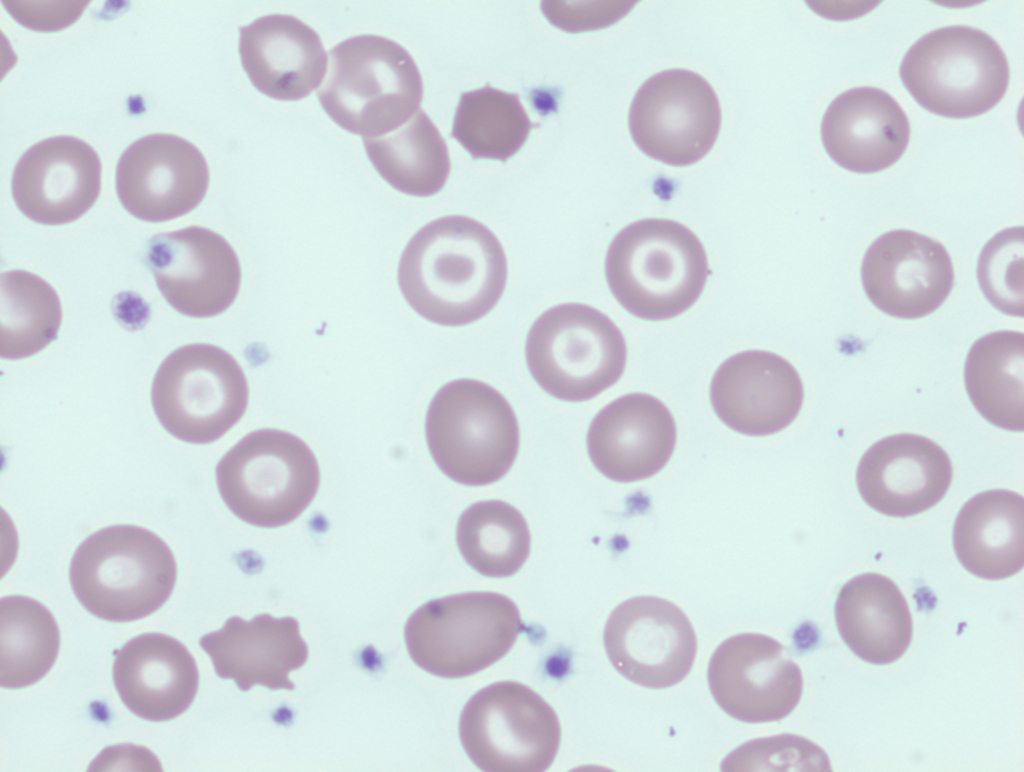
Testing for presence of A and B antigens on red blood cells:
- Forward typing – red blood cells (RBCs) are tested for A and B antigens.
- Reverse typing – serum or plasma is screened for presence of anti-A and anti-B antibodies.
According to a Circular prepared jointly by AABB, the American Red Cross, America’s Blood Centers, and the Armed Services Blood Program:
The ABO group of all red-cell-containing components must be compatible with ABO antibodies in the recipient’s plasma. Serologic compatibility between recipient and donor must be established before any red-cell containing component is transfused. This may be accomplished by performing ABO/Rh typing, antibody screening, and crossmatching by serologic technique or use of a computer crossmatch.

Red cell mass > 25% above that predicted for sex and body mass.

Acquired alpha-thalassemia most commonly develops in patients with myelodysplastic syndrome. It is caused by inactivating somatic mutations of the trans-acting chromatin-associated factor ATRX, which cause dramatic downregulation of alpha-globin gene expression.
Learn more here.
Occurs when there is a reduction in the synthesis of the α-globin chains of adult hemoglobin (HbA, α2β2) relative to beta-globin synthesis. Underproduction of α globin chains gives rise to excess β-like globin chains which form γ4 tetramers, called Hb Bart’s (in fetal life) and β4 tetramers, called HbH (in adult life).
- α thalassemia trait:
- Mutations affecting the α globin genes on one chromosome
- Associated with minimal anaemia
- Compound heterozygotes and some homozygotes for α thalassemia (HbH disease):
- Moderately severe anemia
- Presence of HbH in the peripheral blood
- Hb Bart’s hydrops fetalis syndrome:
- Little or no α globin chains have a very severe form of anaemia
- May cause death in the neonatal period if left untreated

Learn more here.
Acanthocytes are densely stained, spheroidal red blood cells that lack central pallor and have 3-20 irregularly distributed, thorn-like projections of variable size/length/thickness, often with drumstick (knobby) ends. Spicules may occasionally have branches. Typically seen in cirrhosis, but also in hereditary abetalipoproteinemia and related neurological syndromes. Small numbers may be observed post-splenectomy.
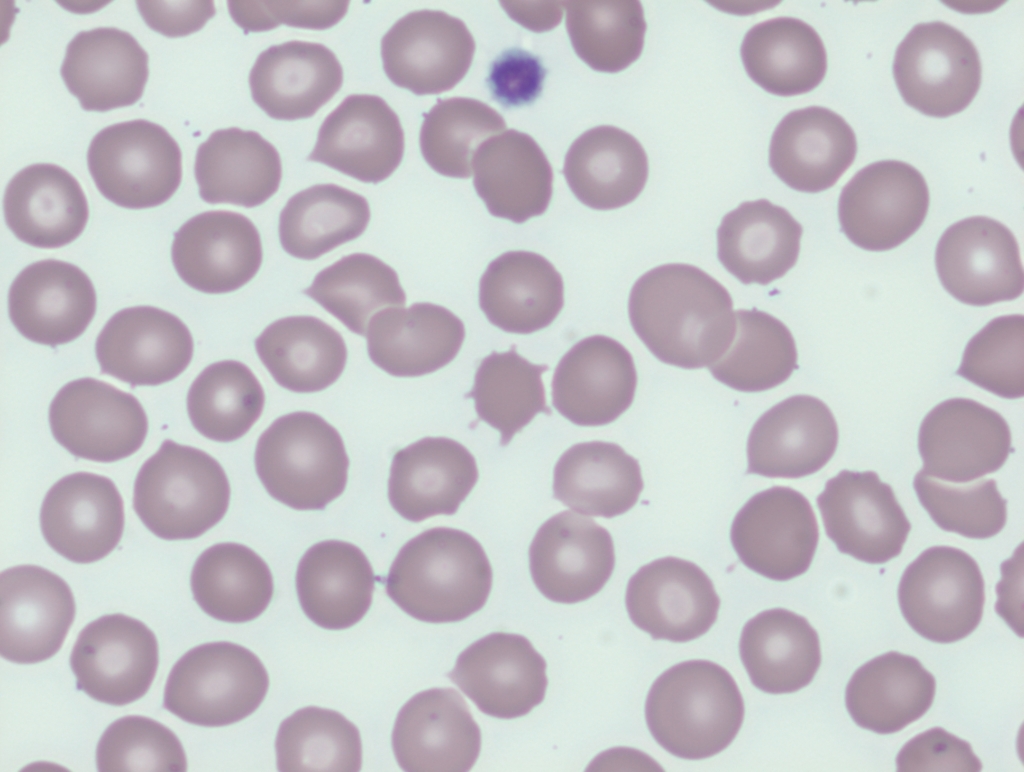
Acute, severe reaction, typically caused by red blood cell ABO incompatibility.
According to the AABB, acute hemolytic transfusion reaction is defined as the rapid destruction of red blood cells during,
immediately after, or within 24 hours of cessation of transfusion. Clinical and laboratory signs of hemolysis are present.
Criteria for diagnosis:
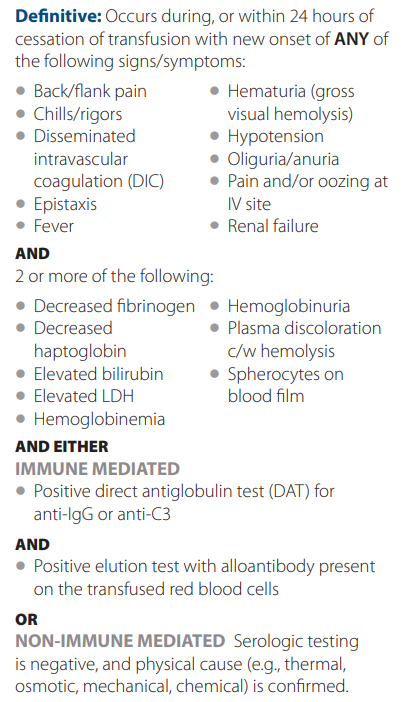

According to the AABB, an allergic reaction is the the result of an interaction of an allergen with preformed antibodies. In some instances, infusion of antibodies from an atopic donor may also be involved. It may present with only muccocutaneous signs and symptoms.
Criteria for diagnosis:

Abrupt reduction in hemoglobin in a patient with sickle cell disease, typically caused by parvovirus B19-mediated suppression of red blood cell production.
ABO compatibility verified using computer system without immediate spin or and antiglobulin crossmatching if significant alloantibodies have not been detected in the recipient sample.
Using AABB guidelines, the electronic crossmatch is possible only if the following conditions are met:
- The patient’s ABO group and Rh type has been done twice and entered in the computer (one group can consist of a record but one must be done on a current in-date specimen). * The computer must alert the technologist if there is a discrepancy between the two groups.
- The donor ABO (and Rh types, if negative) have been confirmed and entered in the computer. The donor’s unit identification number, component name, and ABO/Rh type must also be entered in the computer (manually or by scanning the bar code label on the unit).
- The computer system will alert the technologist to ABO & Rh discrepancies between information on the donor label and results of donor confirmatory testing.
- The computer system will alert the technologist to ensure correct data entry and interpretation, e.g., prevent group O test results from being misinterpreted as group A.
- The computer system will alert the technologist to ABO and Rh discrepancies patient and donor groups. The program should be programed to prevent assigning ABO incompatible blood (e.g., group A red cells to a group O recipient) and to give an alert when assigning Rh-positive red cells to Rh-negative recipients.
Mild-moderate decrease in serum hemoglobin (Hb rarely < 8 g/dL [80 g/L]), associated with acute or chronic inflammation, in which inflammatory mediators lead to increased hepcidin expression, iron sequestration and reduced erythropoiesis. Learn more here.
Defined as increased heterogeneity (or variation) in red blood cell (RBC) size, which can be measured as cell diameter (on a peripheral smear) or cell volume (using the red cell distribution width [RDW] generated by an automated counter).
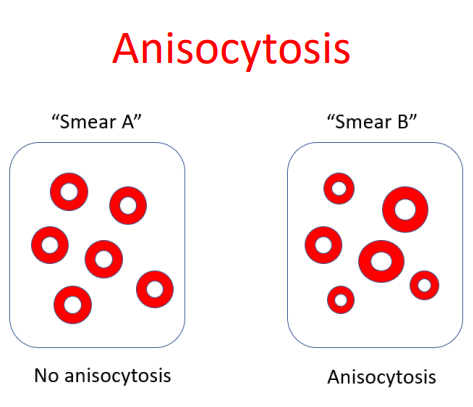
Echinocyte
Drepanocyte
Spur cell
Burr cell
Chronic autoimmune atrophic gastritis
Acanthocyte
Anemia of inflammation
A rare, life-threatening disorder characterized by hypocellular bone marrow resulting in progressive pancytopenia (low reticulocyte, granulocyte, and platelet count) without bone marrow infiltrate, dysplasia or fibrosis. See more here.
A debilitating dermal condition characterized by intense itching, stinging, tingling or burning sensations, without observable
skin lesions, precipitated by contact with water at any temperature. Typically occurs after a shower with warm water. Reported in 5%-69% of patients with polycythemia vera (PV) (41.2% of patients in a recent study of 102 patients with PV). Notably, the onset of aquagenic pruritus precedes diagnosis of PV in almost half of cases.
Congenital secondary erythrocytosis caused by mutations in oxygen sensing/hypoxia-inducible factor (HIF) signaling pathway (autosomal recessive mutations in VHL gene, a negative regulator of hypoxia sensing).
- First hereditary condition of augmented hypoxia sensing to be recognized.
- Autosomal recessive disorder with increased serum erythropoietin levels and hemoglobin concentrations in normoxia.
- Hundreds of patients with Chuvash polycythemia are found in the Chuvash population of central Russia.
- Mutation for Chuvash polycythemia is 598C>T in the von Hippel-Lindau gene (VHL) on chromosome 3p25.
Read more here.
Acquired uncompensated agglutination and premature destruction of red blood cells (RBCs) by autoantibodies (typically IgM) that target RBC antigens with optimal activity between 0 and 4 degrees C (32-39.2 degrees F), but also react at temperatures > 30 degrees C (86 degrees F). Learn more here.
2014 British Committee for Standards in Haematology (BCSH) guideline suggests using serum cobalamin (vitamin B12) cutoff < 148 pmol/L (200 pg/mL) or cutoff derived from local reference range in patient with strong clinical suspicion for vitamin B12 deficiency:

Vitamin B12 concentrations <200 pg/ml in patients with clinical manifestations have a sensitivity of 90%–95% and an estimated specificity of less than 80%. Learn more here.
Rare Diseases NIH: Autosomal dominant (rarely X-linked) inherited blood disorder that leads to anemia and is associated with physical abnormalities such as small head size (microcephaly) characteristic facial features, cleft palate, cleft lip, short and webbed neck, small shoulder blades, and defects of the hands (mostly of the thumbs), as well as defects of the genitalia, urinary tract, eyes and heart. Caused by mutations in several genes. Learn more here.
A sickle cell. Some authors used to refer to sickle cell disease as drepanocytosis (for example, see this 1944 paper).
The EMA binding test uses flow cytometry to determine the amount of fluorescence (reflecting EMA bound to specific transmembrane proteins) derived from individual red cells. Red blood cells (RBCs) are incubated with EMA dye, which covalently binds to band 3 and other proteins on RBC surface. The mean fluorescence of EMA-stained RBCs in patients with hereditary spherocytosis is lower when compared with control RBCs due to the decreased amount of target proteins. The EMA binding test is easy to use, and test results are available for reporting in 2–3 h.
Read more here.
An increase in the number of red blood cells (RBCs) relative to the plasma volume. It is manifested by a persistent increase in the venous hematocrit:
- Relative erythrocytosis – elevated hematocrit associated with normal red cell mass, resulting from contracted plasma volume most often due to dehydration.
- Absolute erythrocytosis:
- An increased red cell mass above 125% of the predicted value for the body mass of the patient.
- Red cell mass is rarely measured; hemoglobin and/or hematocrit typically used as surrogates for red cell mass.
A vasomotor disturbance characterized by periodic attacks with triad of increased temperature, erythema, and burning pain resulting from hyperperfusion of skin areas, especially in the feet and hand. Typically relieved by cooling and aggravated by warming. May be primary or secondary. Causes of secondary erythromelalgia include polycythemia vera and essential thrombocythemia.

Learn more here.
A glycoprotein hormone secreted by peritubular cells in the kidney upon detection of reduced oxygen in the circulation. Binds to the erythropoietin receptor present on erythroid precursor cells in the bone marrow, resulting in stimulation of red blood cell production.
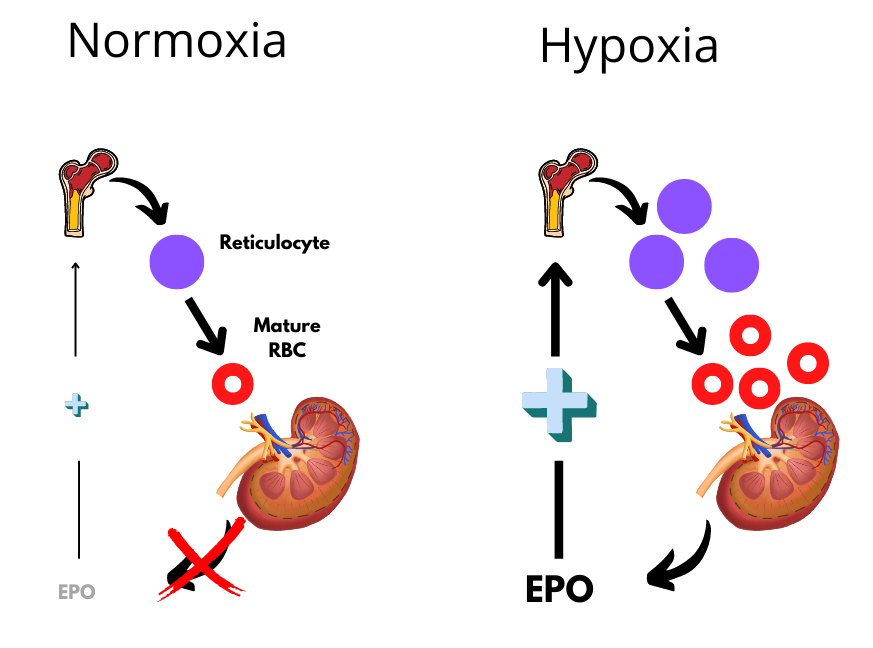
Combined immune thrombocytopenia (ITP) and autoimmune hemolytic anemia. Learn more here.
Acute hemolytic anemia resulting from ingestion of fava beans. Learn more here.
Eculizumab or ravulizumab (C5 inhibitors). Learn more here.
Fluorescent aerolysin, used in flow cytometry to diagnose paroxysmal nocturnal hemoglobinuria (PNH) (selectively binds the GPI anchor with high affinity).
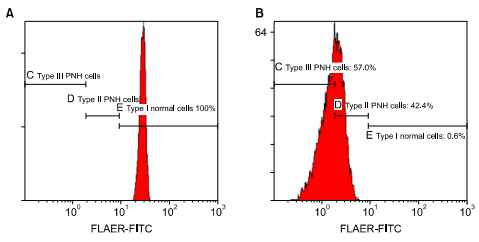
A condition in which body iron stores are normal or increased, but iron incorporation into red cell precursors is impaired. One example is anemia of inflammation.
An X-linked genetic disorder caused by mutations in G6PD gene resulting in reduced activity of the G6PD enzyme. Learn more here.
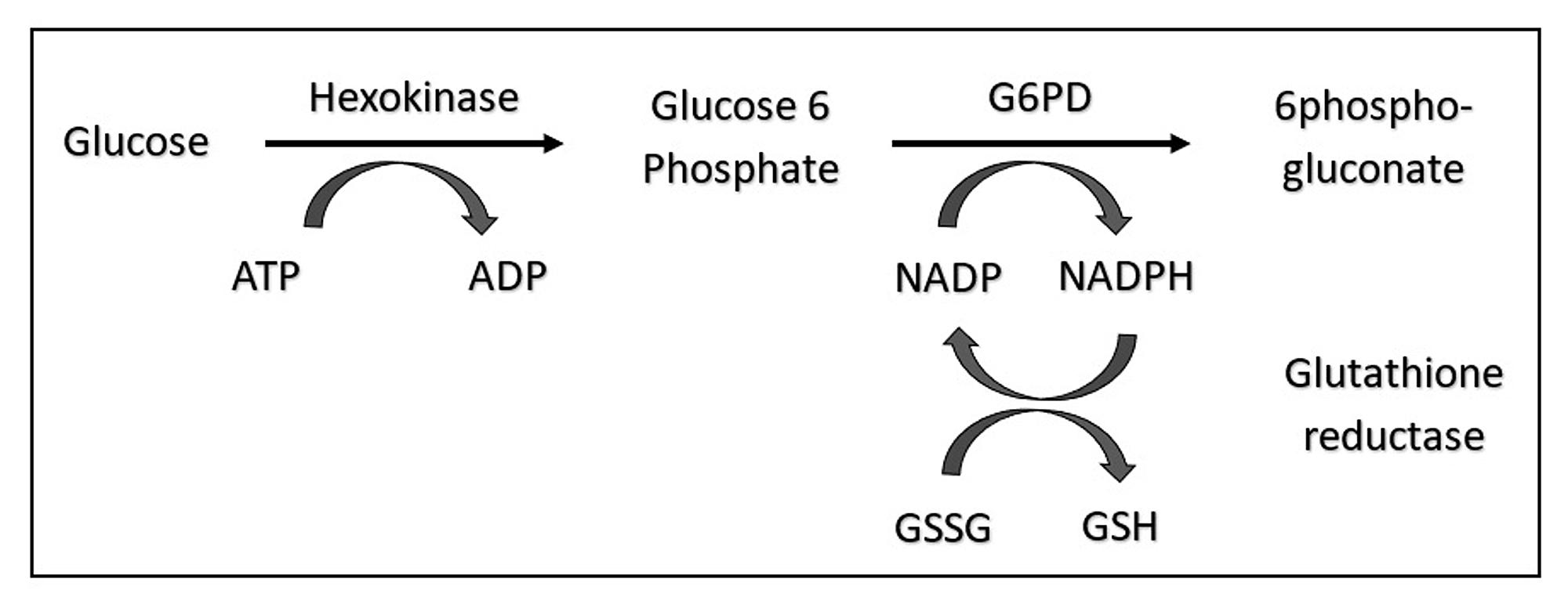
HbH is comprised of β4 tetramers made from excess β globin chains in alpha thalassemia.
Learn more here.
A 25-amino acid peptide hormone produced by liver hepatocytes in response to circulating iron levels which inhibits iron absorption from the intestinal mucosal cells and release by macrophages and hepatocytes through degradation of ferroportin-1. Hepcidin plays a critical role in iron absorption and recycling. Learn more here.
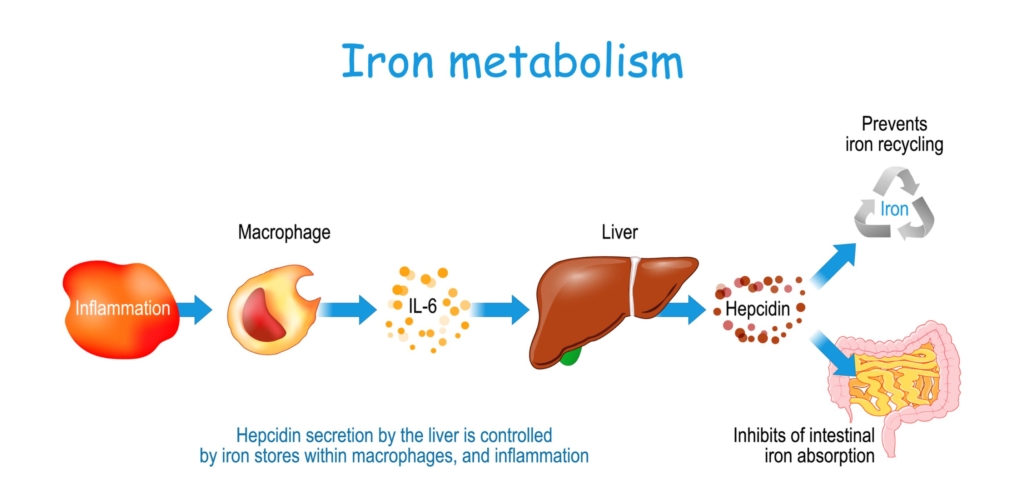
Hydroxyurea is a ribonucleotide reductase inhibitor
Refers to the inability to produce adequate number of red blood cells in the presence of increased immature erythroid precursors.
Per Cazzola, M.:
- In ineffective erythropoiesis, the erythroid marrow is active and expanded (erythroid hyperplasia) but its efficacy in terms of red cell production is impaired and may lead to anemia.
- Erythroblasts are predominant in the bone marrow, and the M/E ratio is <1:1.
- Ineffective erythropoiesis is a major pathogenetic mechanism that is responsible for anemia in several inherited and acquired disorders, including:
- Thalassemia:
- Transfusion-dependent β-thalassemia (also known as β-thalassemia major)
- Nontransfusion-dependent β-thalassemia (also known as β-thalassemia intermedia; including β-thalassemia/Hb E)
- Inherited sideroblastic anemias
- Congenital dyserythropoietic anemias
- Megaloblastic anemias
- Myelodysplastic syndrome
- Thalassemia:
- Patients with ineffective erythropoiesis may have evidence of parenchymal iron overload, which derives from suppression of hepcidin production.
Lactate dehydrogenase (LDH) is a ubiquitous enzyme that catalyzes the conversion of lactate to pyruvate and back, resulting in conversion of NAD⁺ to NADH and back. LDH exists in five distinct forms, named LDH-1 through LDH-5, each having differential expression in different tissues:
- LDH-1 is found primarily in heart muscle.
- LDH-2 is the major isozyme of the reticuloendothelial system and red blood cells.
- LDH-3 is highest in the lung.
- LDH-4 is highest in the kidney, placenta, and pancreas.
- LDH-5 is highest in the liver and skeletal muscle.
Meets the criteria for diagnosis polycythemia vera despite lower-than-threshold hemoglobin/Hct.
Hemolytic anemia + schistocytes on the peripheral smear caused by high shear environment from partially occluded vessels or paravalvular jet streams (in the latter case, the term MAHA is used even though a paravalvular leak does not involve the microvasculature).
Free, unbound iron in the plasma. Consists of multiple chemical structures of iron (including labile plasma iron) associated with other plasma components. Typically appear when transferrin saturation > 70%. Rapidly taken up by parenchymal cells of the liver, heart, anterior pituitary cells and pancreas, where they generate reactive oxygen species and cause organ damage.
Learn more here.
Monitors the extent of red blood cell (RBC) shape change (elongation index) within a range of osmotic gradient under known shear stress and is a measure of hydration status of RBCs. Used to diagnose RBC membrane disorders.
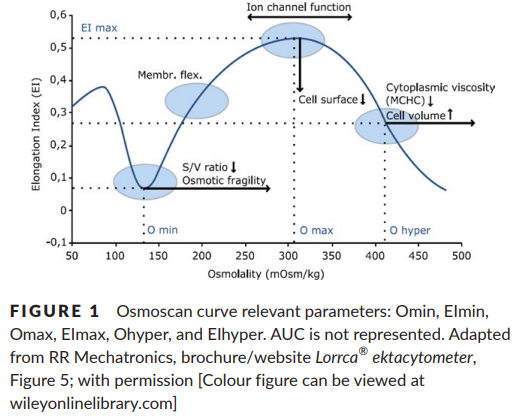
Read more here.
A form of pica, defined by eating of at least a tray of ice daily for 2 months or of ice chips. Accounts for over 50% of pica in patients with iron deficiency. Learn more here.
A chronic myeloproliferative neoplasm associated with increased risk of thrombosis and bleeding, and disease progression to either myelofibrosis and/or acute leukemia. Caused by somatic mutations in JAK2 (Janus kinase), typically V617F.
Porphyrias are a group of eight panethnic inherited metabolic disorders of heme biosynthesis. Each results
from a specific enzymatic alteration in the heme biosynthesis pathway.
Learn more here.
Hematocrit > 51% lasting > 1 month post transplantation (usually develops within the first year post transplant). Occurs in 10%-15% of renal transplant recipients and is usually self-limiting. Most common after kidney transplant, but may also occur after other types of transplant, including hematopoietic stem cell transplantation. Learn more here.
A type of absolute erythrocytosis (increased red cell mass) caused by an intrinsic defect in the stem cell population leading to
autonomous proliferation of red cell progenitors within the bone marrow. There are two types of primary erythrocytosis:
- Congenital erythrocytosis (primary familial or congenital polycythemia) caused by gain of function mutations in the erythropoietin receptor.
- Acquired erythrocytosis (polycythemia vera) caused by a somatic mutation in the Jak2 gene.
A syndrome characterized by isolated normocytic normochromic anemia with severe reticulocytopenia and significant reduction or absence of erythroid precursors in bone marrow. Learn more here.
A trace byproduct of the last step in heme synthesis. In iron deficiency, zinc, instead of iron, is incorporated into the protoporphyrin ring, resulting in increased zinc protoporphyrin levels (iron protoporphyrin is also called heme). ZPP may be used as an assay for iron deficiency.
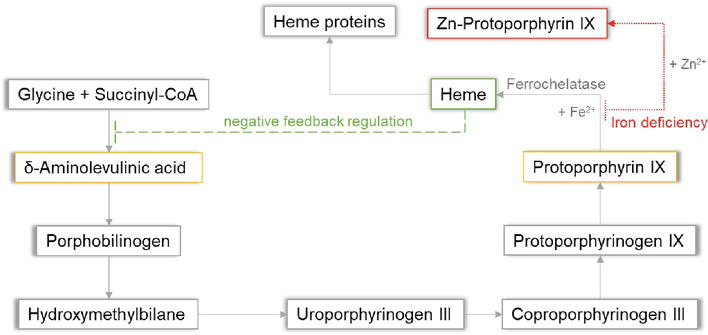
Elevated hematocrit associated with normal red cell mass resulting from contracted plasma volume most often due to dehydration.
- Absence or presence of PNH clone
- Clone size in red blood cells:
- Total PNH clone size
- Percentages of Type II and type III cells
- PNH clone size in white blood cells:
- Neutrophils
- Monocytes
Learn more here.
Sleep-related sensorimotor disorder characterized by an urge to move that occurs during rest or is exacerbated by rest, occurs in the evening or night and disappears or improves with movement. The pathophysiology is unknown.
Learn more here.
Rouleaux is the arrangement of >3 red cells in a linear of branched pattern resembling a stack of counts. Seen in conditions associated with an inflammatory response and acute phase reaction or multiple myeloma.
Elevated red cell mass (absolute erythrocytosis) caused by a factor external from the bone marrow, typically erythropoietin (other factors include testosterone and cortisol). Learn more here.
A very small fraction of total ferritin circulates in blood. In contrast to its cellular counterpart, serum ferritin does not contain any iron and is almost entirely made up of L chains. It does not play a role in iron transport or cellular iron uptake. Indeed, its function is unknown. Serum ferritin is widely used as a convenient surrogate measure of body iron stores.
A group of chronic hemolytic anemias characterized by the presence of at least one hemoglobin S allele (HbS; p.Glu6Val in HBB) and a second HBB pathogenic variant leading to excessive production of hemoglobin S (HbS). Learn more here.
Hemolytic uremic syndrome caused by Shiga toxin-producing Escherichia coli (STEC).
Learn more here.
Screening for complement-activating alloantibodies in a patient who will receive a blood transfusion, using commercially available group O RBCs that express all antigens as required by the FDA.
Per the AABB:
Pretransfusion compatibility testing begins with the type and screen procedure. The recipient’s ABO group and Rh type are determined first; then a screening procedure is used to detect any unexpected non-ABO blood group antibodies that may be present. If the screening test reveals the presence of an antibody, the specificity of that antibody is determined by an antibody identification panel. Once the specificity of the antibody has been identified, donor units of the appropriate ABO group and Rh type are screened for the corresponding antigen. Units that are negative for that antigen are crossmatched with the recipient to ensure compatibility.
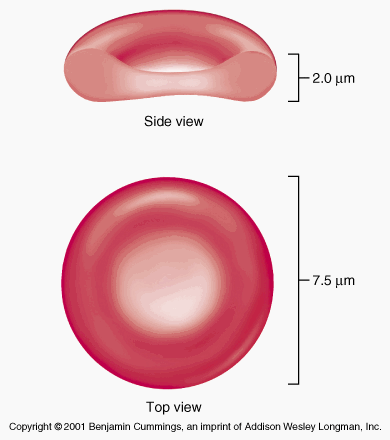
Caused by inactivating somatic mutations of the trans-acting chromatin-associated factor ATRX, which cause dramatic downregulation of alpha-globin gene expression.
Learn more here.
Partial deficiency of the third enzyme of heme synthesis, porphobilinogen deaminase (or hydroxymethylbilane synthase). AIP is an autosomal dominant condition.
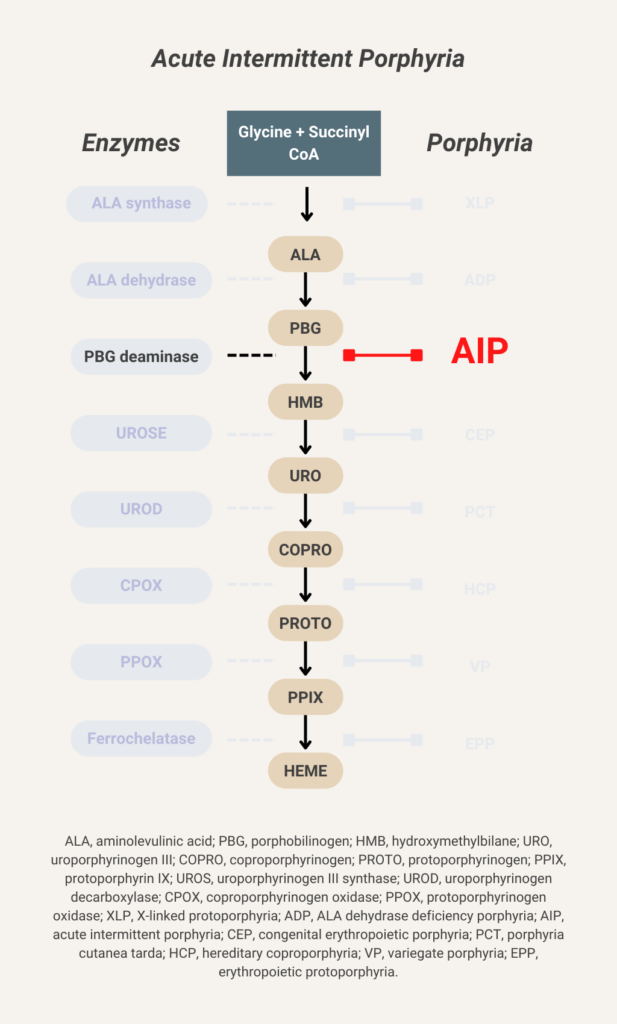
Autosomal dominant mutations of one of the following genes:
- SPTA1 encoding alpha-spectrin
- SPTB encoding beta-spectrin
- EPB41 encoding protein 4.1
Autosomal recessive mutations in one of the following genes:
- SPTA1 (encoding alpha-spectrin)
- SPTB (encoding beta-spectrin)
- EPB41 (encoding protein 4.1)
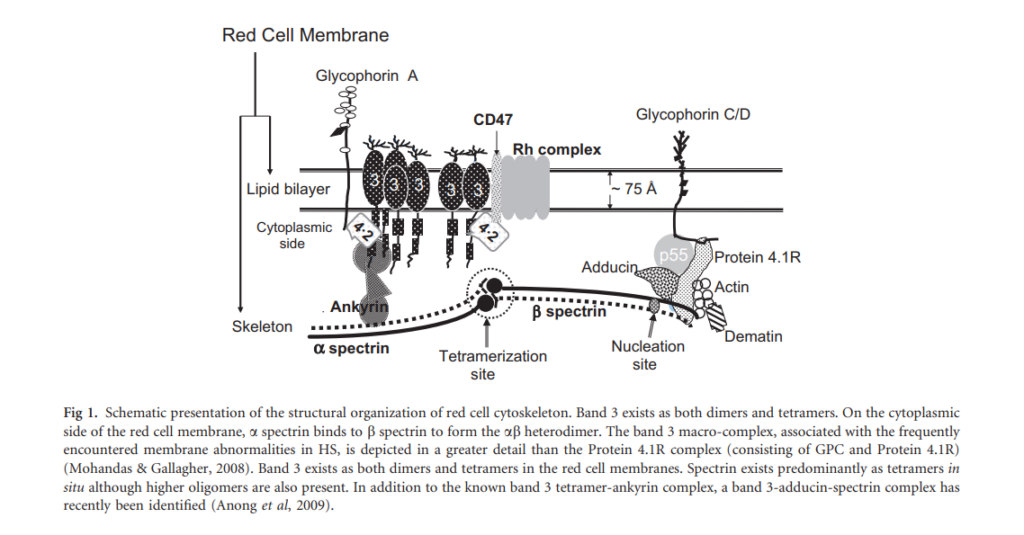
Autosomal dominant (75% of cases) or autosomal recessive (25%) mutations in genes that encode red blood cell (RBC) membrane proteins, resulting in qualitative defects of the encoded proteins. Genes with mutations include:
- ANK1 encoding ankyrin
- SLC4A1 encoding band 3
- SPTA1 encoding alpha-spectrin
- SPTB encoding beta-spectrin
- EPB42 encoding protein 4.2
Mutations results in reduced binding of spectrin cytoskeleton to lipid bilayer of RBCs, leading to reduced membrane surface area (reduced surface area-to-volume ratio), reduced RBC deformability and increased osmotic fragility.
Autoimmune-mediated atrophic gastritis resulting from the destruction of gastric parietal cells and leading to deficient production of intrinsic factor (necessary for vitamin B12 absorption), and reduced vitamin B12 absorption. The immune response is directed against the gastric H/K–ATPase, which accounts for associated achlorhydria.
See more here.
Somatic mutations in JAK2 (Janus kinase), specifically V617F in exon 14 (reported in about 95% of patients) and mutations in exon 12 (reported in about 4% of patients), leading to constitutive activation of Jak2 signaling in hematopoietic stem cells.
Congenital gain-of-function mutations in erythropoietin receptor (EPOR)
Autoantibody, typically IgG, directed against red blood cell self-antigens with maximal reactivity at 37 degrees C (98.6 degrees F). Learn more here.
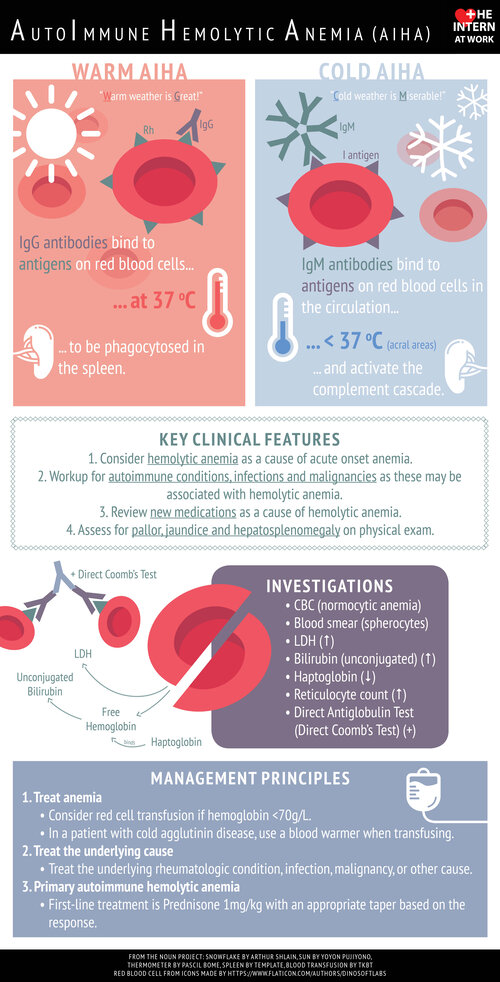
- Thrombocytopenia,
- Microangiopathic hemolytic anaemia
- Damage to various organs, predominantly the kidney and the brain
Learn more here.
- Hemolytic anemia
- Bone marrow failure
- Thrombosis
The common feature to all forms of hemolytic uremic syndrome is the presence of endothelial cell lesions in the microvasculature of the kidney and, less frequently, of other organs… all forms of hemolytic uremic syndrome share a final common procoagulant and proinflammatory phenotype of activated endothelial cells.
Learn more here.
Receptor in the terminal ileum that binds and internalizes the vitamin B12-intrinsic factor complex. Learn more here.
Based on spectrophotometric measurement of cyanide derivative of hemoglobin.
Protocol includes:
- Blood is mixed with a solution containing potassium cyanide and potassium ferricyanide (Drabkin’s solution).
- The red blood cells are lysed, producing an evenly disturbed hemoglobin solution.
- Potassium ferricyanide transforms hemoglobin into methemoglobin, and methemoglobin combines with potassium cyanide to produce hemiglobincyanide (cyanmethemoglobin).
- Cyanmethemoglobin has a peak absorbance at 540 nm and is measured photometrically.
- The absorbance is compared with that of the standard hemiglobincyanide solution by using a formula to obtain the amount of hemoglobin.
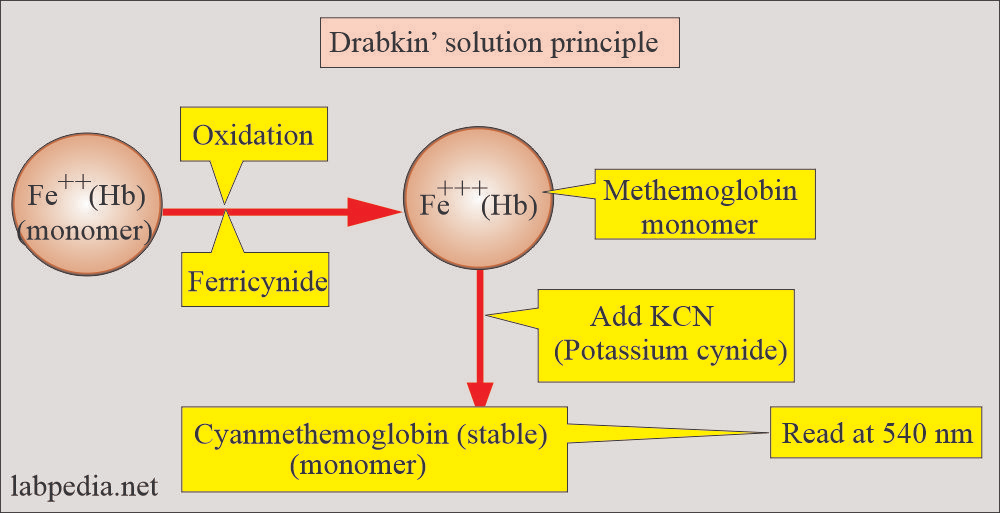
Hb<13 g/dL in men, Hb < 12 g/dL in women (<11 g/dL if pregnant)
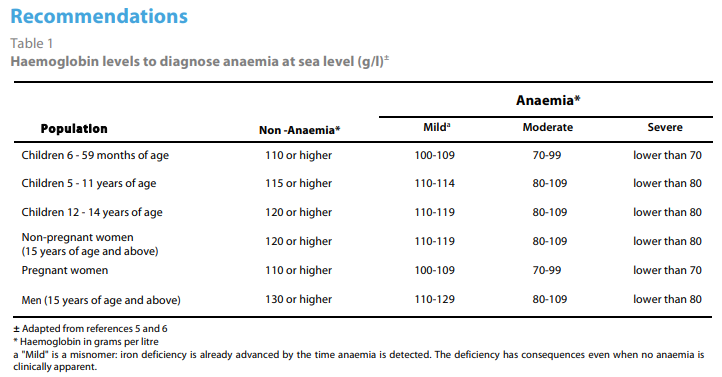
Although the terms are often used interchangeably, they have slightly different meanings.
Erythrocytosis is an increase in red blood cells (RBCs) relative to the volume of blood. Primary erythrocytosis is associated with an elevated red cell mass (a change in the numerator in RBC/volume), while relative erythrocytosis is not (rather, it is a change in the denominator in RBC/volume). An important caveat is that an increased number of microcytic RBCs, for example in thalassemia, may be associated with a normal or even low red cell mass (Hct=MVC x RBC count). Indeed, before its cause was known thalassemia was historically referred to as microcytic erythrocytosis.
Polycythemia refers to increased red cell mass, manifested by increased Hct and/or hemoglobin, and usually caused by increased RBC count (erythrocytosis). In theory, an elevated MCV could produce increased red cell mass independent of RBC number (Hct=MVC x RBC count). As the name ‘poly’ implies, polycythemia more accurately refers to pan-myeloproliferation, but this definition is not normally adhered to.
Hb measures the total weight of hemoglobin contained within a given volume of blood. It is reported in g/dl or g/L. The Hct is the fractional volume of blood that is comprised of red cell cells. The Hct is expressed as a percentage.
A polychromatophilic cell is a red cell that is slightly larger than a mature red cell whose cytoplasm has a bluish tinge when observed on a normally stained peripheral smear. Most if not all of these cells correspond to reticulocytes, which are RNA containing red cells that are detected either with intravital stains (where blood is not fixed, but rather incubated alive with stain) or FACS analysis using an RNA fluorochrome. Reticulocyte staining is more sensitive than Wright-Giemsa staining for detecting reticulocytes: all true polychromatic cells are reticulocytes, whereas not all reticulocytes would be detectable as polychromatic cells.
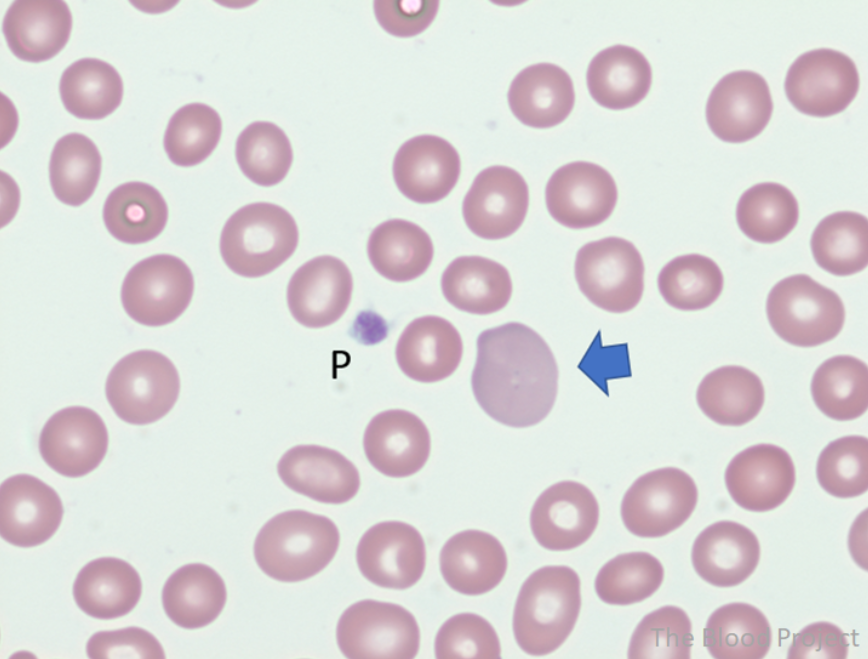
Sickle cell anemia is a subset of sickle disease that includes the 2 most severe forms: homozygous sickle cell disease (HbSS), sickle beta0 thalassemia.
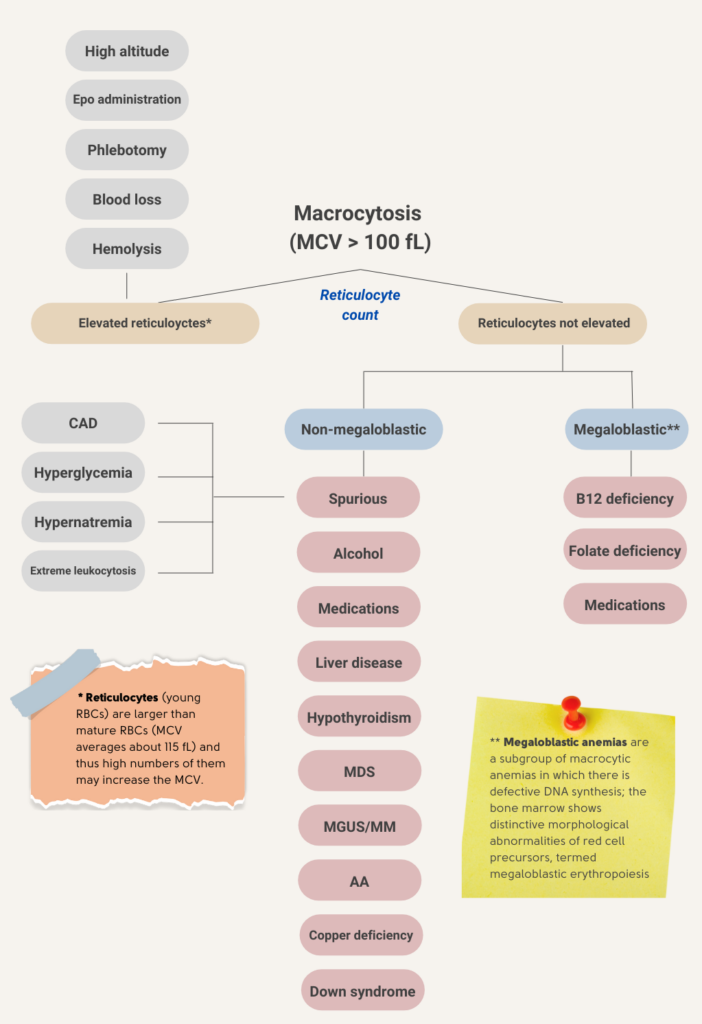
1000 μg vitamin B12 daily. Learn more here.
1-4 mg/kg/day IV for 3-14 days based on clinical signs; do not exceed 6 mg/kg/day. Learn more here.
Erythrocyte sedimentation rate is the distance red cells fall by gravity in a vertical tube of anticoagulated whole blood. Learn more here.
Each unit of RBCs contains enough hemoglobin to increase the hemoglobin concentration in an average-sized adult by approximately 1 gram/deciliter (g/dL) (increase hematocrit by 3%). Learn more here.
Examination of urine for excess porphobilinogen (measurement of 5-aminolaevulinic acid is not essential to establish the diagnosis). Elevated porphobilinogen levels in urine (or plasma) are specific for acute porphyria. The test can be performed in a random sample with the result normalized per gram of urine creatinine; a 24-hour collection is not required.
Urinary porphobilinogen and 5-aminolaevulinic acid are increased in all three acute hepatic porphyrias (acute intermittent porphyria, hereditary coproporphyria, and variegate porphyria) although the concentrations are higher and longer lasting in acute intermittent porphyria than in the other two types (hereditary coporphyria and variegate porphyria).
With a recorded porphobilinogen over excretion (>10 times the upper limit), treatment can be started immediately, with further laboratory investigations used to define the porphyria type in the proband.
For diagnosis of the type of acute porphyria in the proband, plasma fluorescence emission spectroscopy is a fi rst-line test because a peak at 624–628 nm establishes the diagnosis of variegate porphyria.
Learn more here.
Involved in the biosynthesis of glycosylphosphatidylinositol (GPI) anchors, including CD55 and CD59.
Global anemia prevalence in 2010 was 32.9%, causing 68.36 (95% uncertainty interval [UI], 40.98 to 107.54) million years lived with disability (8.8% of total for all conditions [95% UI, 6.3% to 11.7%]). In 2013 nearly 2 billion people or 27% of the world’s population were affected.
Learn more here.
Typically to maintain HbS level at < 30% immediately prior to next transfusion.
Fraction of whole blood that is made of RBCs (also known as packed cell volume).
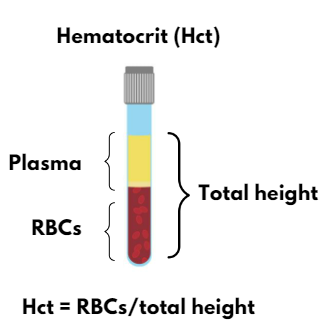
Inheritance of AIP, VP and HCP is autosomal dominant.
Jak2 V617F mutation is a G to T somatic mutation at nucleotide 1,849 in exon 14 of JAK2, leading to constitutive activation of signaling pathways downstream of the erythropoietin receptor. It occurs in 95% of patients with polycythemia vera as well as a smaller percentage of patients with other myeloproliferative neoplasms, encodes a constitutively active Janus kinase that upregulates JAK/STAT signal transduction, resulting in unregulated myeloproliferation (erythrocytosis, often with leukocytosis and/or thrombocytosis).
May occur in:
- Polycythemia vera (95% of cases)
- Essential thrombocythemia (50-60% of cases)
- Primary myelofibrosis (50-60% of cases)
- Refractory anemia with ringed sideroblasts associated with significant thrombocytosis (50% of cases)
Learn more here.
Meningococcal meningitis, which occurred in two of 100 patients included in prospective trials. Patients receiving eculizumab should receive specific meningococcal prophylaxis.
Learn more here.
Represents mean weight of hemoglobin (Hb) per red blood cell (RBC); calculated by dividing hemoglobin (Hb) by the RBC count (RBC).
MCH = Hb/RBC

Measures concentration of hemoglobin (Hb) per unit volume of red blood cells (RBCs); calculated by dividing hemoglobin (Hb) by hematocrit (Hct).
MCHC = Hb/Hct
Under normal conditions:
330 g/L = 150 g/L/0.45

Defined as the value that describes the average size of red blood cells in a blood sample. Individual red blood cells may be microcytic, normocytic or macrocytic.
Normal MCV is 80-100 fL.
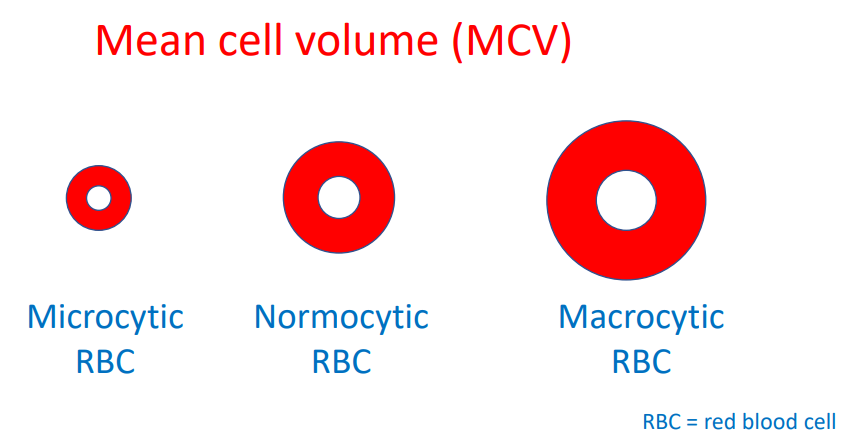
Crizanlizumab is a monoclonal antibody medication that binds to P-selectin, inhibiting white blood cell-endothelial and platelet-endothelial interactions on the blood vessel wall.
Overproduction of neurotoxic heme precursor from the liver caused by deficient enzyme in heme biosynthesis pathway, often in conjunction with precipitating factor that increases rate of heme synthesis. Learn more here.
Jak2 mutation (V617F)
Iron deficiency anemia, reported to account for about 50% of cases.
Vaso-occlusive crisis Learn more here.
Menstrual blood loss.
Blood loss from the gastrointestinal tract.
Hereditary spherocytosis; reported prevalence from 1 in 1,000 to 1 in 3,000 persons.
Babesia microti
At steady state, about 1%-2% of circulating red blood cells (RBCs) are reticulocytes, corresponding to an absolute reticulocyte count of approximately 25-100 × 109/L.
Measures red blood cell (RBC) lysis incubated in solution of varying concentrations of sodium chloride. RBC lysis is measured by detection of light absorbance by free hemoglobin immediately after sampling and after 24-hour incubation at 37 degrees C (98.6 degrees F). Osmotically fragile cells, such as spherocytes, lyse at relatively lower saline concentrations compared to normal RBCs.
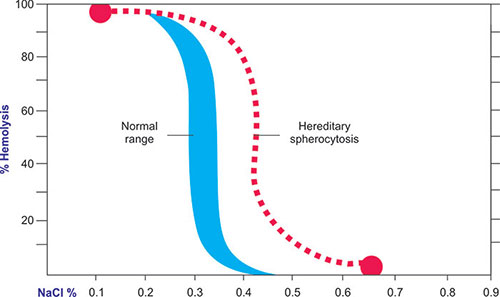
Hypoxia-inducible factor 1-alpha (HIF1-alpha)
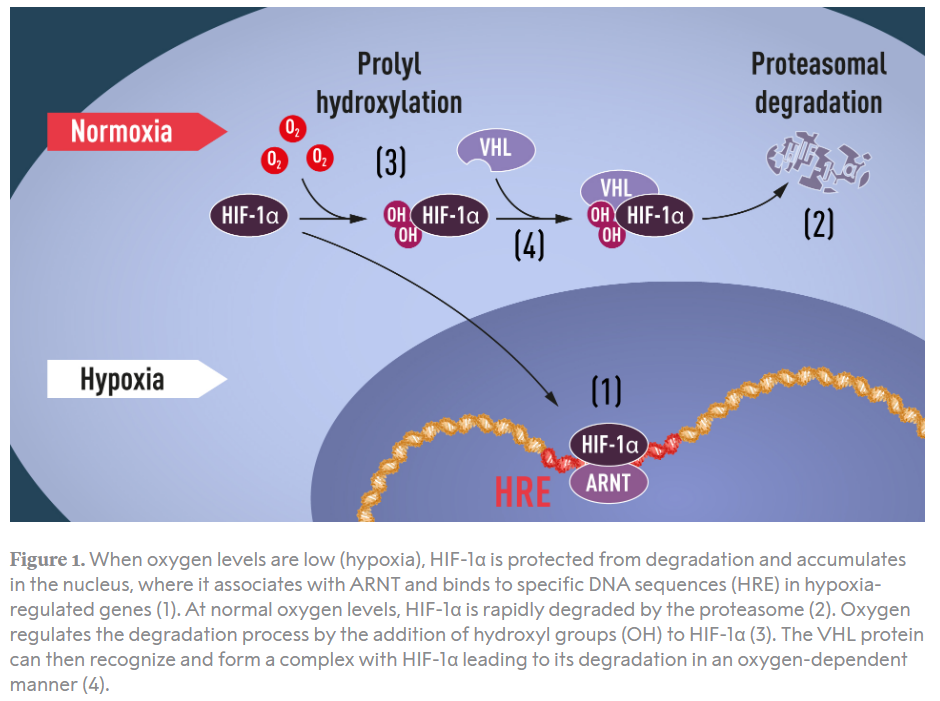
Measures proportion of RBCs that have a cellular hemoglobin weight of < 17 pg (analogous to the mean corpuscular hemoglobin, not mean corpuscular concentration – in other words, the term hypochromic is misleading). The parameter is available in some automated hematology analyzers. May be helpful for the assessment of iron availability (absolute or functional deficiency) for erythropoiesis.
A rare disorder associated with triad of:
- Postcricoid dysphagia which is:
- Typically painless, intermittent or progressive over years, and limited to solids
- Occasionally associated with weight loss
- Iron deficiency anemia
- Upper esophageal webs
Other features:
- Most of the patients are white middle-aged women, in the fourth to seventh decade of life.
- The dysphagia is usually painless and intermittent or progressive over years, limited to solids and sometimes associated with weight loss.
- Reported to be a risk factor for developing squamous cell carcinoma of the upper gastrointestinal tract.
Learn more here.
Anemia of inflammation (the anemia is not always chronic in nature).
0.01%-0.1%
Estimated to affect about one in 75,000 people in European countries, apart from in northern Sweden, where, because of a founder effect, it is more frequent (one in 1000).
Learn more here.
5.4% in the United States, up to 80% in resource-limited countries. Learn more here.
Reported prevalence of anemia from all causes 33%-49% in patients with bariatric procedures. Learn more here.
- Mild anemia (Hb 9.5–10.9 g/dl) – 30%
- Moderate anemia (Hb 8.0 and 9.4 g/dl) – 9%
- Severe anemia (Hb 6.5–7.9 g/dl) – 1%
Learn more here.
12%-24% reported prevalence of anemia from all causes in patients with IBD. Learn more here.
0·23–0·42 cases per million population. Learn more here.
Iron deficiency reported to occur in > 30% of patients after 5 years from surgery, with similar rate after Roux-en-Y gastric bypass (RYGB) and sleeve gastrectomy. Learn more here.
Reported prevalence of iron deficiency in pregnancy 18%:
- 7% in the first trimester
- 30% in the third trimester
Learn more here.
Reported prevalence of iron deficiency approximately 1% in male adults and ≥ 11% in female adults. Learn more here.
Macrocytosis occurs in about 3% of the general population.
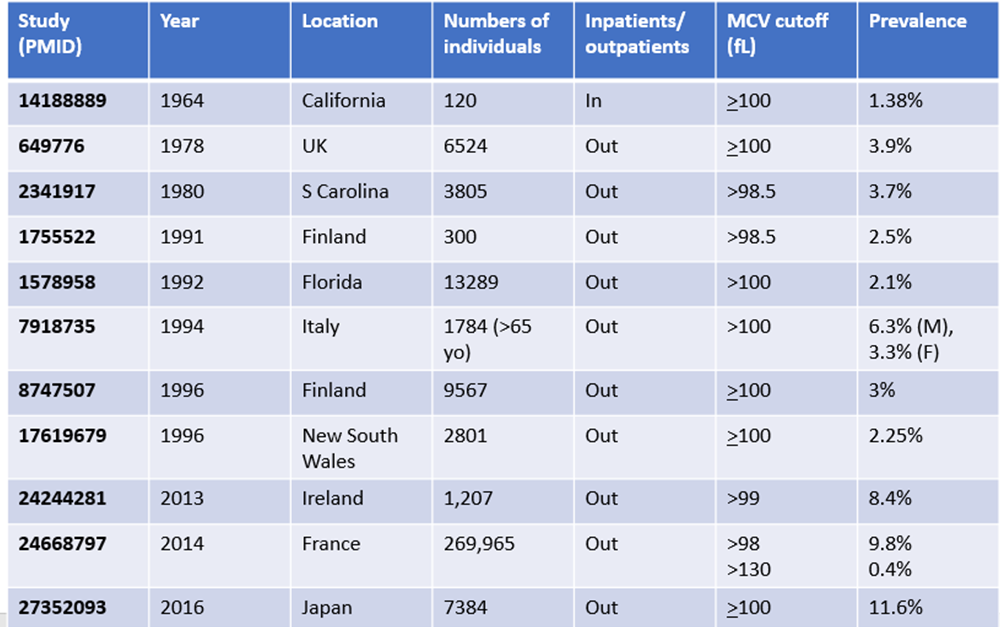
About 20%. Learn more here.
30-45% of patients
RBC transfusion at Hb level of < 7 g/dL (70 g/L) with a target Hb of 7-9 g/dL (70-90 g/L). Transfusion trigger should not exceed 90 g/L in most critically ill patients.
AABB recommendations in 2016:


Consider treatment of symptomatic anemia with red cell transfusion to minimize symptoms; transfusion usually required when Hb is < 6 g/dL (60 g/L).
For patients having orthopedic or cardiac surgery and are hospitalized and hemodynamically stable, transfuse at Hb level ≤ 8 g/dL (80 g/L).
AABB recommendations in 2016:
0.1 % to 0.3 % per year. A nearly 7-fold significantly higher relative risk of gastric cancer in patients with vs without pernicious anemia.
Learn more here.
0.4–0.7% per year. Learn more here.
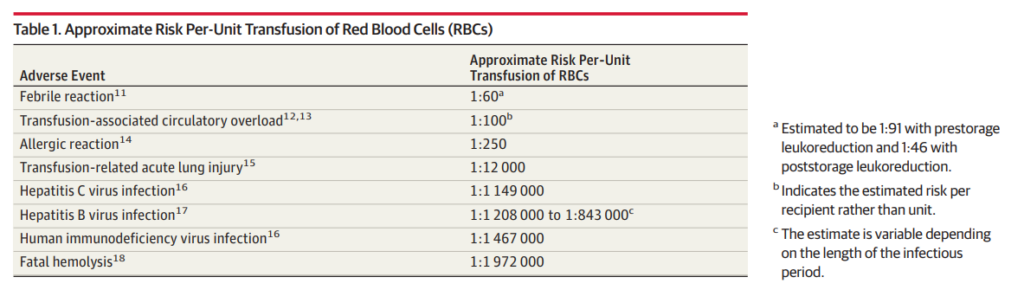
Acute chest syndrome
Anemia of inflammation
> 24 hours after transfusion.
Within 24 hours of transfusion or during transfusion.
Eculizumab, a monoclonal anti-C5 antibody that blocks the entry of C5 into C5 convertase.
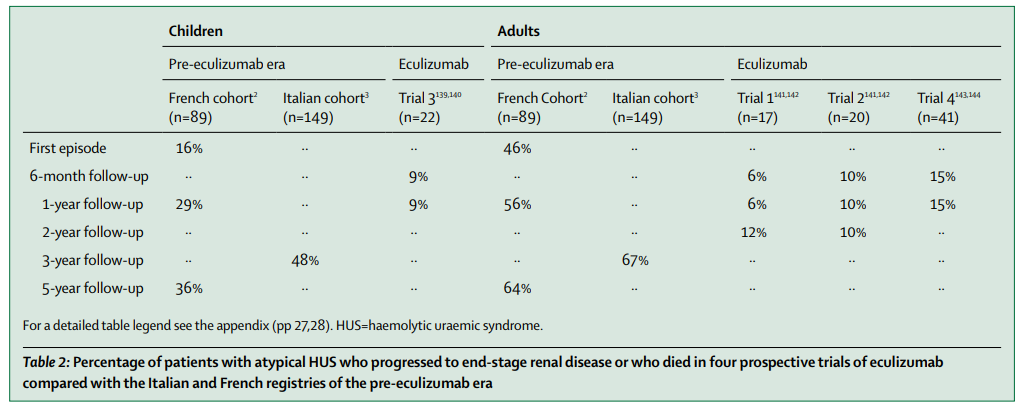
Learn more here.
- Prodromic phase including minor behavioral changes such as:
- Anxiety
- Restlessness
- Insomnia
- Severe abdominal pain, but this pain might also be felt in the back or thighs.
- Nausea, vomiting, and constipation are common.
- Symptoms of increased sympathetic activity, including:
- Tachycardia
- Excess sweating
- Hypertension
Physical exam usually normal.
Labs may show hyponatremia.
Urine may be red or dark-colored.
Most acute attacks last for no longer than 1 or 2 weeks.

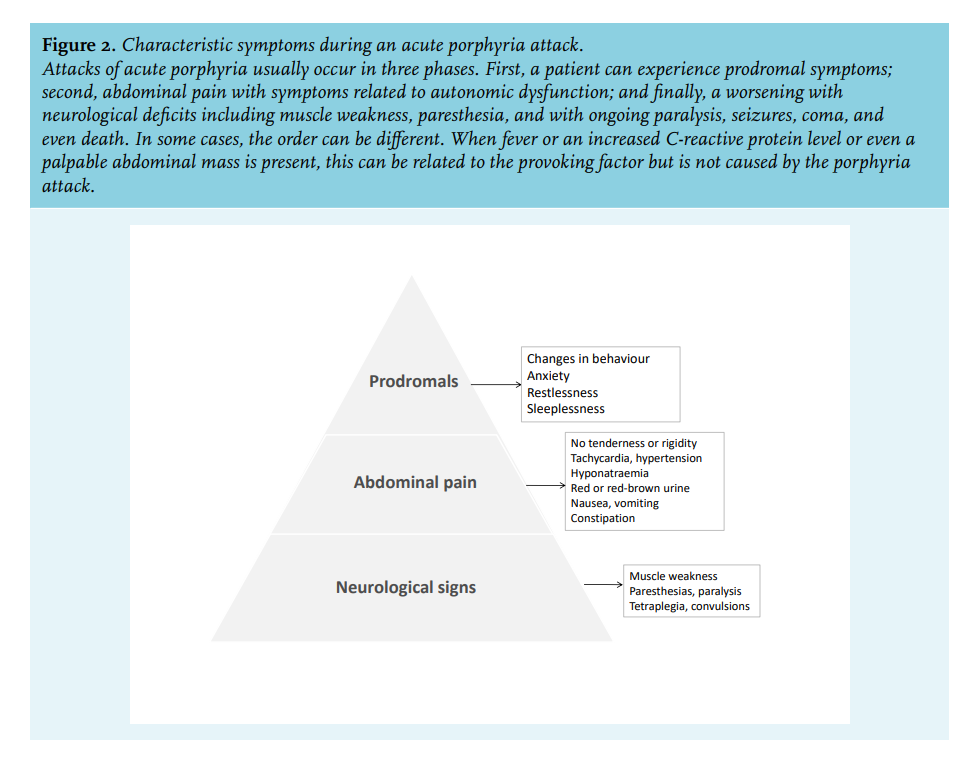
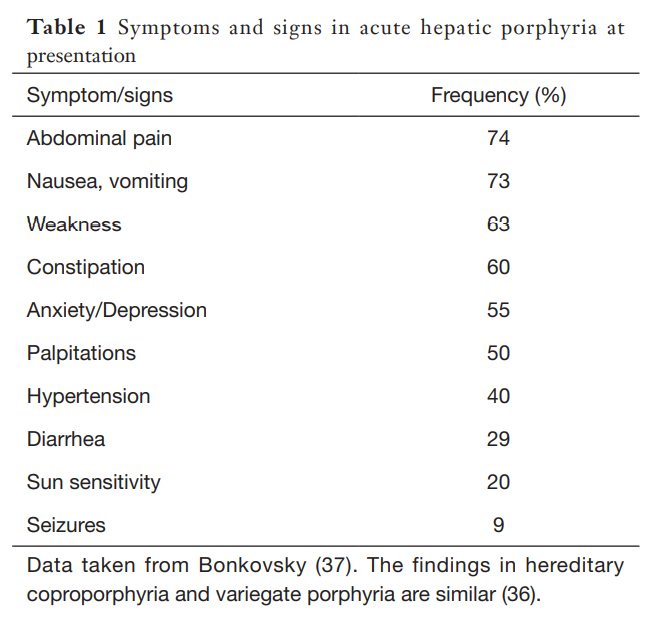
Learn more here.
300 mL to 400 mL
According to a Circular prepared jointly by AABB, the American Red Cross, America’s Blood Centers, and the Armed Services Blood Program:
Depending upon the collection system used, a single whole blood donation typically contains either 450 mL (+/- 10%) or 500 mL (+/- 10%) of blood collected from allogeneic blood donors. After plasma is removed, the resulting component is RBCs, which has a hematocrit between 65% to 80% and a usual volume between 225 mL and 350 mL. Red Blood Cells additive solutions (AS) may be mixed with the red cells remaining after removal of nearly all of the plasma to extend the shelf life (see Table 2). The typical hematocrit of AS RBCs is 55% to 65%, and the volume is approximately 300 to 400 mL.
Acute pulmonary edema due to volume overload from transfusion. Usual onset within 1-2 hours of transfusion. Risk factors include transfusion of large volumes of components or high flow rates. Treatment is supportive and may include lasix, and rarely therapeutic phlebotomy.
According to Nareg Roubinian, TACO is pulmonary edema primarily related to circulatory overload with criteria developed by the National Healthcare Safety Network including 3 or more of the following within 6 hours of transfusion:
- Acute respiratory distress
- Radiographic pulmonary edema
- Elevated central venous pressure
- Evidence of left heart failure
- Elevated B-type natriuretic peptide (BNP)
- Positive fluid balance
According to the AABB, TACO is defined as new onset or exacerbation of 3 or more of the following within 12 hours of cessation of transfusion:
- Elevated brain natriuretic peptide (BNP) or NT-pro BNP relevant biomarker.
- Evidence of cardiovascular system changes not explained by underlying medical condition (Elevated central venous pressure, evidence of left heart failure including development of tachycardia, hypertension, widened pulse pressure, jugular venous distension, enlarged cardiac silhouette and/or peripheral edema).
- Evidence of fluid overload.
- At least 1 of the following:
- Evidence of acute or worsening respiratory distress (dyspnea, tachypnoea, cyanosis and decreased oxygen saturation values in the absence of other specific causes) and/or,
- Radiographic or clinical evidence of acute or worsening pulmonary edema (crackles on lung auscultation, orthopnea, cough, a third heart sound and pinkish frothy sputum in severe cases).
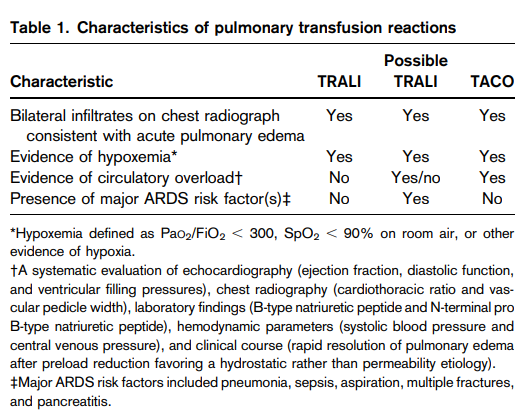
Form of acute lung injury caused by donor leukocyte antibodies (occasionally in recipient) and other leukocyte activating agents in plasma-containing components including whole blood, RBCs, platelets, cryoprecipitate, and fresh frozen plasma. Symptoms arise within 6 hours of transfusion and typically resolve after 48-96 hours. Treatment is supportive.
The Canadian Consensus Criteria defines TRALI as acute pulmonary edema after transfusion in the absence of circulatory overload or alternate acute respiratory distress syndrome (ARDS) risk factors:


According to the AABB, TRALI is defined as:
- No evidence of acute lung injury (ALI) prior to transfusion, and
- ALI onset during or within 6 hours of cessation of transfusion, and
- Radiographic evidence of bilateral infiltrates, and
- No evidence of left atrial hypertension (i.e., circulatory overload), and
- Hypoxemia defined by any of these methods:
- PaO2/FiO2 less than or equal to 300 mmHg
- Oxygen saturation less than 90% on room air
- Other clinical evidence
Increased iron intake sustained over a period of time as a result of multiple red blood cell (RBC) transfusions which may be exacerbated by increased absorption through the gastrointestinal tract.
Hct by microhematocrit centrifugation is slightly higher owing to the effect of trapped plasma.
Unexplained anemia of the elderly (UAE) is an entity characterized by a hypoproliferative normocytic anemia that is not due to the nutritional deficiency, chronic kidney disease or inflammatory disease, and in which the erythropoietin response to anemia is blunted.
Learn more here.
First-line testing:
- Complete blood count
- Peripheral blood smear
- Renal function
- Liver function
- Arterial oxygen saturation
- Carboxyhemoglobin
- Serum ferritin
- Serum erythropoietin:
- A low erythropoietin level (< 2.9 mU/mL) specific (92%) and moderately sensitive (64%) for the diagnosis of polycythemia vera.
- A high erythropoietin level (> 15.1 mU/mL) specific (98%) but had poor sensitivity (47%) for the diagnosis of secondary erythrocytosis.
- Jak2 mutational analysis (peripheral blood) – based on the pretest probability of polycythemia vera (PV):
- In the primary care setting, where the probability of PV is low, clinical evaluation for secondary causes of erythrocytosis paired with a high erythropoietin level can rule out PV in most patients.
- In hematology clinics, where the probability of PV is higher, erythropoietin level and JAK2 V617F mutation testing are done concurrently.
- Patients with a low or normal erythropoietin level and no JAK2 V617F mutation are further evaluated with JAK2 exon 12 mutation testing (on peripheral blood or marrow aspirate, based on local practice) and a bone marrow biopsy.
Second-line testing:
- When no diagnosis is made, selected patients with onset of erythrocytosis at a young age or compatible family history should undergo testing for high-oxygen-affinity hemoglobins, and gene sequencing for mutations involving the erythropoietin receptor or oxygen-sensing pathways.
- Investigations for secondary erythrocytosis should be symptom directed and may include:
- Chest radiography
- Overnight oximetry for suspected sleep apnea
- Pulmonary function tests for hypoxic lung disease
- Venous blood gas sampling (carboxyhemoglobin level)
- Echocardiography to rule out right to left cardiac shunting
- Abdominal–pelvic imaging can help exclude an erythropoietin-producing tumor or conditions associated with local renal hypoxia.
- Neuroimaging to rule out meningioma or cerebellar hemangioblastoma should be ordered for patients with unexplained neurologic symptoms as these tumors have been associated with autonomous erythropoietin production.
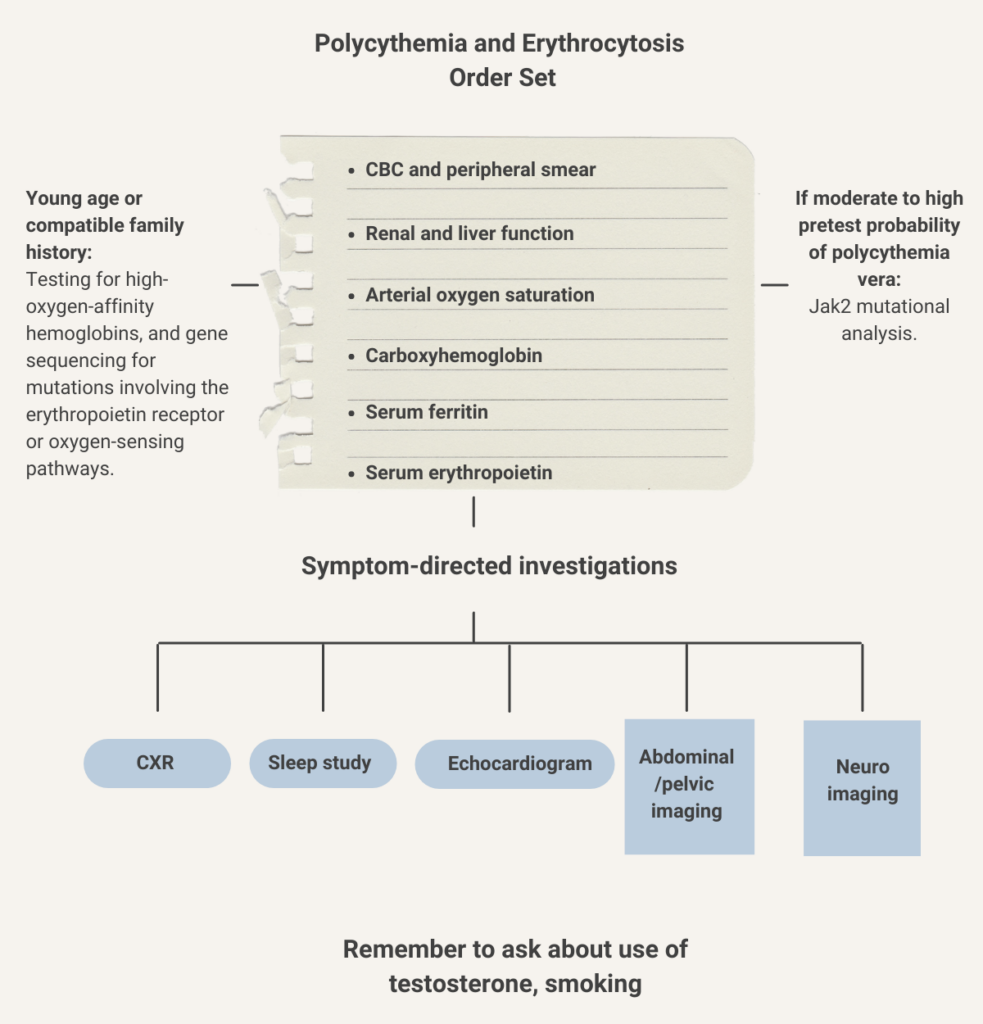
Learn more here.
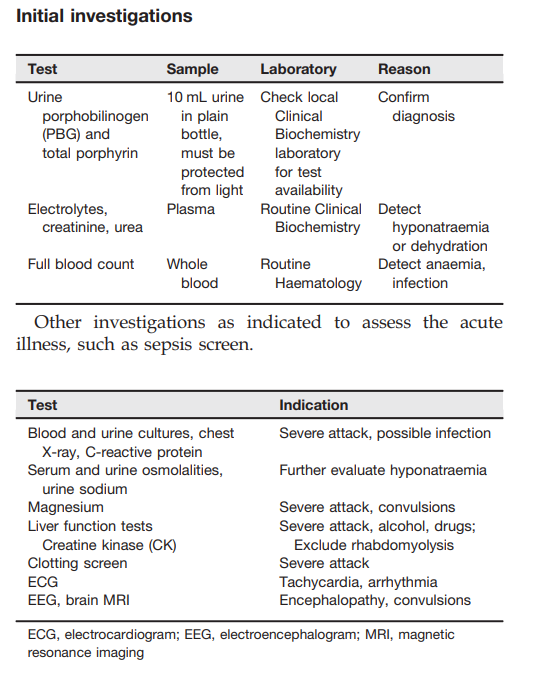
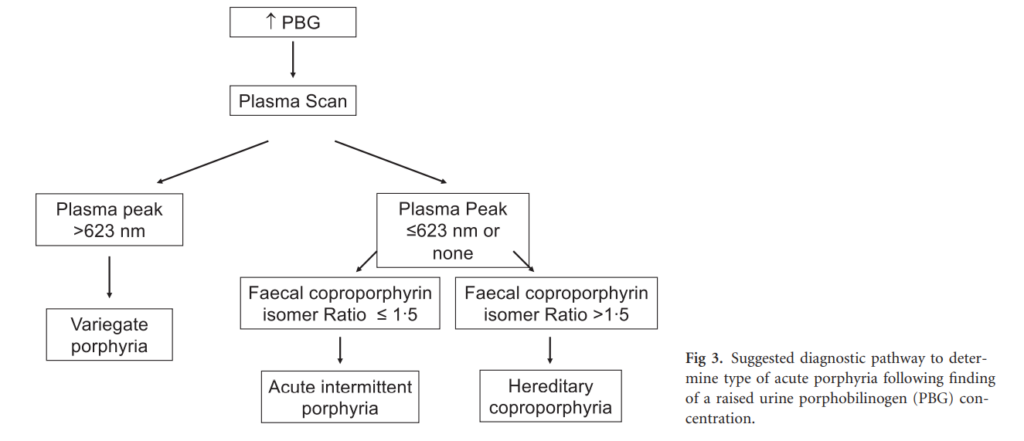
- Electrical impedance (Coulter principle)
- Optical light measurements
- Optical absorbance
- Optical light scatter
- Fluorescence
- Cyanmethemoglobin measurement of hemoglobin
About 70-80%
Reported mean daily iron absorption 6% for male adults and 13% for nonpregnant female adults in their childbearing years. Amounts to 1-2 mg/day (increasing to 6 mg in the third trimester of pregnancy).
Renal failure requires prompt initiation of dialysis in more than 50% of cases regardless of the cause of hemolytic uremic syndrome, and in more than 75% of adults with atypical hemolytic uremic syndrome.
Learn more here.
40–60% of patients.
Learn more here.
10%-20% of patients. Learn more here.
>90% of such patients have evidence for a clonal expansion of kappa-positive B cells and a monoclonal IgM paraprotein. Learn more here.
About 25%-35%. Learn more here.
Iron deficiency is estimated to account for about 50% of all cases of anemia in pregnant and nonpregnant female adults.
IgM, reacting optimally between 0 and 4 degrees C. Learn more here.
Homozygous sickle cell disease (HbSS) and sickle beta0 thalassemia. .
Both markers are highly sensitive, but MMA is more specific (for example, homocysteine levels may also be increased in patients with folate deficiency). MMA values considered high vary from 0.28 to 0.75 mcmol/L.
2014 British Committee for Standards in Haematology guidelines for the diagnosis and treatment of cobalamin and
folate disorders:


Learn more here.
Intrinsic factor antibodies are more specific (98-99% vs. 50%) but less sensitive (40-60% vs. 80-90%) than antiparietal antibodies.
Humans have no mechanisms to excrete iron. Iron is normally lost through:
- Skin desquamation
- Sweat – sweat contains up to 0.3 mg iron/L
- Gut
- Menstruation
- Pregnancy
Terminal ileum.
Anti-parietal cell antibodies, which target the gastric enzyme H+/K+ATPase proton pump. Learn more here.
Louis Diamond and Kenneth Blackfan, who first described the disorder in 1938 (Diamond LK, Blackfan KD. Hypoplastic Anemia. American Journal of Diseases of Children. 1938;56:464–467).

About 90% of cases reported in female adults, mean age at diagnosis of 47 years.
Robert S. Evans, first author on paper (titled “Primary thrombocytopenic purpura and acquired hemolytic anemia: evidence for a common etiology”) that first described the syndrome in 1951.
V. Koneti Rao wrote:
Known to his colleagues and friends as Bud Evans, he was a native Seattleite. Unfortunately, Evans was killed in a tragic car accident on Mercer Island in 1974 at the young age of 62.
Henry Stanley Plummer (1874–1936) and Porter Paisley Vinson (1890–1959) who were physicians on the staff of the Mayo Clinic. Learn more here.
Because megaloblastic red cell precursors are destroyed in the bone marrow (ineffective erythropoiesis) and these immature cells contain hemoglobin, LDH and AST.
Why are polychromatophilic cells bluish in color on Wright-Giemsa-stained peripheral blood smears?
Because of residual RNA

Lymphoma
About 30%
Unexplained fever > 38 degrees C (100.4 degrees F) for ≥ 3 days, weight loss of over 10% body weight in prior 6 months, and drenching night sweats.
Diffuse large B-cell lymphoma
SLL is a different manifestation of the same disease as CLL, with the main difference being that abnormal lymphocytes mainly accumulate in lymph nodes and other tissues, with no evidence of cytopenias or bone marrow involvement. 1
About one-third of patients
Plasma cell disrders
Monoclonal gammopathy (M spike) with monoclonal (M) protein in serum < 3 g/dL, bone marrow clonal plasma cells < 10%, and absence of related organ or tissue impairment (no end-organ damage, including anemia, hypercalcemia, renal insufficiency, or bone lesions) or amyloidosis related to the plasma cell dyscrasia.
Serum M-protein ≥ 15 g/L, non-IgG subtype, and abnormal FLC ratio.
- Increased calcium (serum calcium > 1 mg/dL [0.25 mmol/L]
- Renal insufficiency (creatinine clearance < 40 mL/minute or serum creatinine level > 2 mg/dL [177 mcmol/L])
- Anemia (hemoglobin > 20 g/L [2 g/dL] below lower limit of normal or hemoglobin < 100 g/L [10 g/dL])
- Bone lesions (≥ 1 osteolytic lesions on skeletal radiography, computed tomography [CT], or positron emission tomography [PET]-CT)
More than 1 of: IgG or IgA monoclonal protein ≥ 3g/dL, urinary monoclonal protein ≥ 500 mg per 24 hours, and clonal bone marrow plasma cell 10%-60% in the absence of myeloma defining events or amyloidosis.
Monoclonal gammopathy of clinical significance
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of unknown significance
Serum protein electrophoresis
Serum protein electrophoresis (SPEP) is a test that measures the amount of heavy chain monoclonal protein made by myeloma cells. SPEP separates all the proteins in the blood according to their electrical charge. Densitometry is then used to determine the height of the various peaks, resulting in a graph of the results, as shown in various examples below.
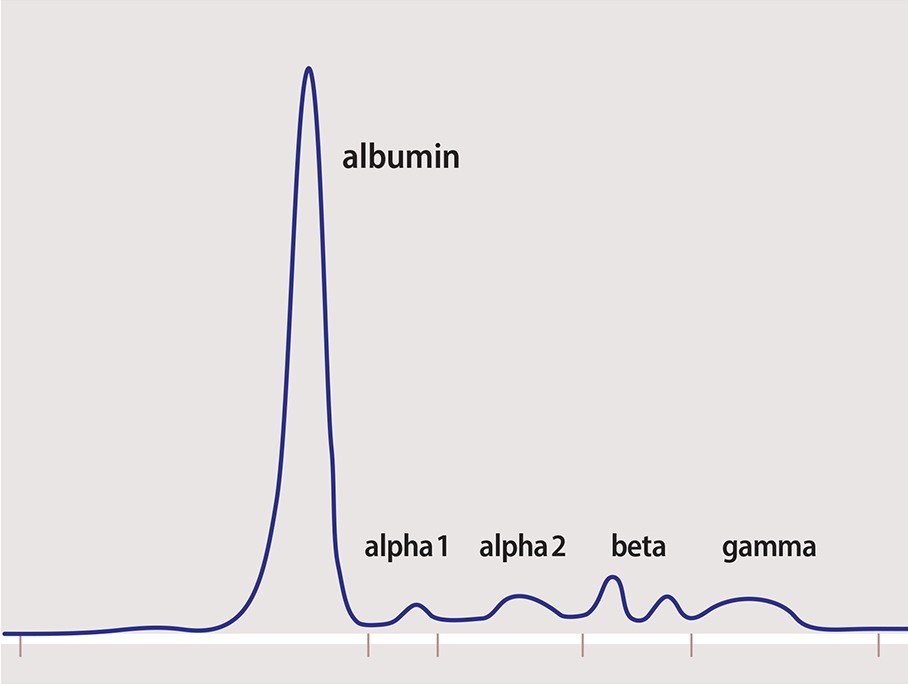
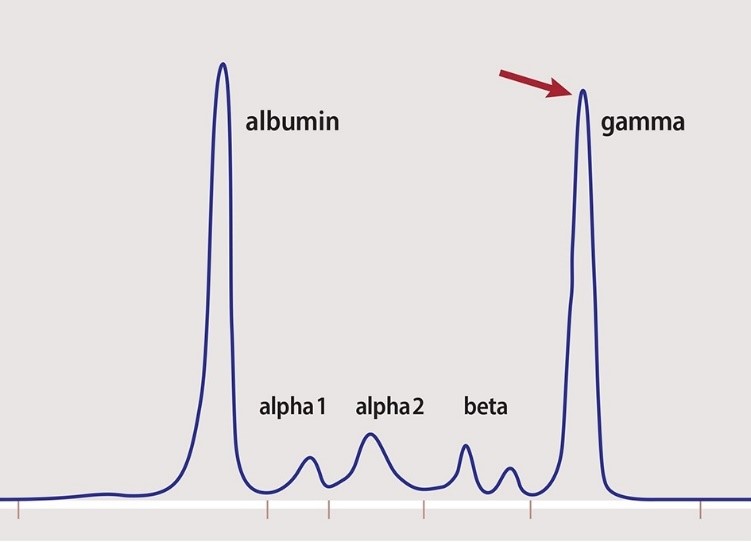
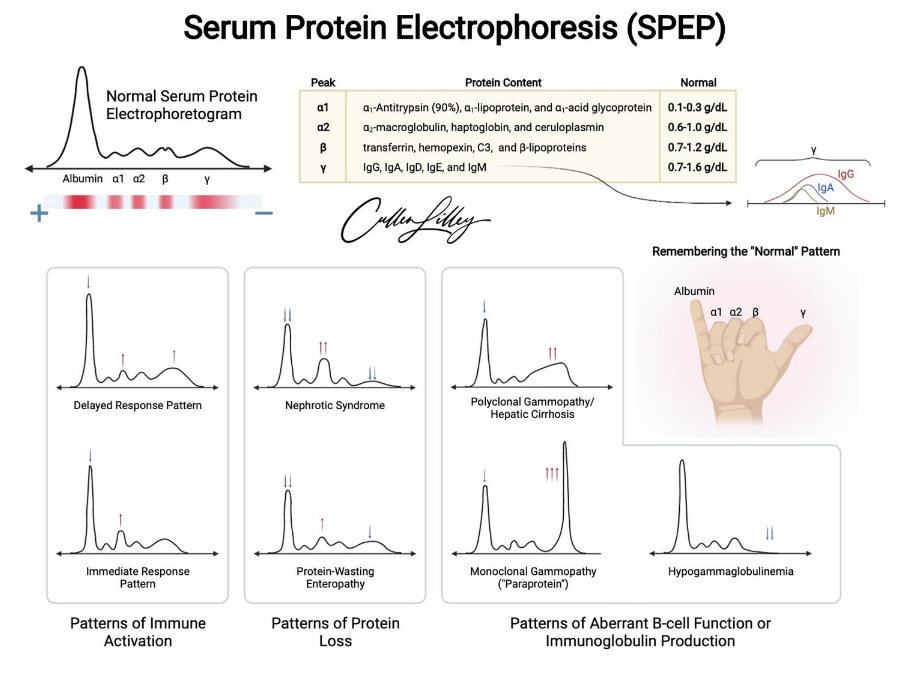
Paraproteins produced by proliferation of single clone of plasma cells or B lymphocytes that appear as a narrow band or spike (M spike) on serum electrophoresis. Usually IgG, but may be IgM, IgA or IgD.
About 1% per year
Iron overload
No. In patients homozygous for C282Y genotype, 25% of men reported to have disease manifestations compared to only 1% of women.
Yes. But they account for <5% of cases of hereditary hemochromatosis. A list of these conditions can be found here (from 2019 ACG Clinical Guideline)
They result in hepcidin deficiency and decreased degradation of ferroportin. The resulting elevation in ferroportin levels causes increased net dietary iron absorption in the intestine and increased export of iron via ferroportin from liver and splenic macrophages and hepatocytes into the blood.
About 4,500
1-2 mg is derived from intestinal absorption.
Each unit of RBCs (processed from 420 mL of donor blood) contains about 200 mg of iron (0.47 mg iron/mL of whole donor blood or 1.08 mg iron/mL of pure RBCs).
The human body normally contains 40 to 50 mg/kg of iron, which amounts to 3-4 grams. It is distributed in the following compartments:
- Hemoglobin in RBCs – about 30 mg/kg
- Myoglobin in muscle – about 4 mg/kg
- Iron-containing enzymes – about 2 mg/kg
- Storage in ferritin and hemosiderin – 0-2,000 mg
- Plasma transferrin – 1-2 mg
About 25 mg of iron daily (far more than the 1 mg that is absorbed by the gastrointestinal track).
Yes, according to 2019 ACG Clinical Guideline:

Hereditary hemochromatosis, other causes of iron overload (transfusional and non-transfusional), alcohol, inflammation, liver disease, hemophagocytic lymphohistiocytosis (HLH), cancer, hereditary hyperferritinemia/cataract syndrome. Learn more here.
Primary cause: Hereditary hemochromatosis
Secondary causes:
- Iron-loading anemias
- Parenteral iron overload
- Chronic liver disease
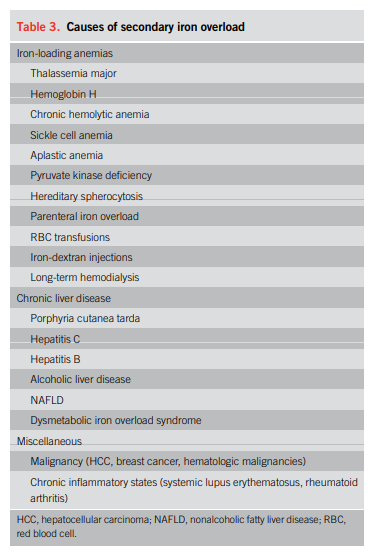
Mutations in:
- Hepcidin
- Hemojuvelin
- Transferrin receptor 2
- Ferroportin
Classification of hereditary hemochromatosis:
| Classification | Genes involved | Inheritance | Protein function | Clinical manifestations |
|---|---|---|---|---|
| Type 1A HH (homozygote) | Mutations in HFE: C282Y | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 1B HH (compound heterozygote) | Mutations in HFE: C282Y H63D | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 1C HH | Mutations in HFE: S65C | AR | Possible elevations in serum iron/ ferritin, no evidence of tissue iron deposition. | |
| Type 2A juvenile HH | HJV (hemojuvelin) | AR | Involved in hepcidin synthesis, BMP co-receptor. | Earlier onset, ,30 years old, hypogonadism and cardiomyopathy are prevalent. |
| Type 2B juvenile HH | HAMP (hepcidin) | AR | Downregulation of iron efflux from erythrocytes. | Earlier onset, ,30 years old, hypogonadism and cardiomyopathy are prevalent. |
| Type 3 HH | TFR2 (transferrin receptor 2) | AR | Involved in hepcidin synthesis, interaction with transferrin. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 4A HH (FPN disease) | SLC40A1 (FPN) | AD | Duodenal iron export. | Iron deposition in the spleen is very common, lower tolerance to phlebotomies and may have anemia. |
| Type 4B HH (nonclassical FPN disease) | SLC40A1 (FPN) | AD | Resistance to hepcidin. | Fatigue, joint pain. |
Learn more here.
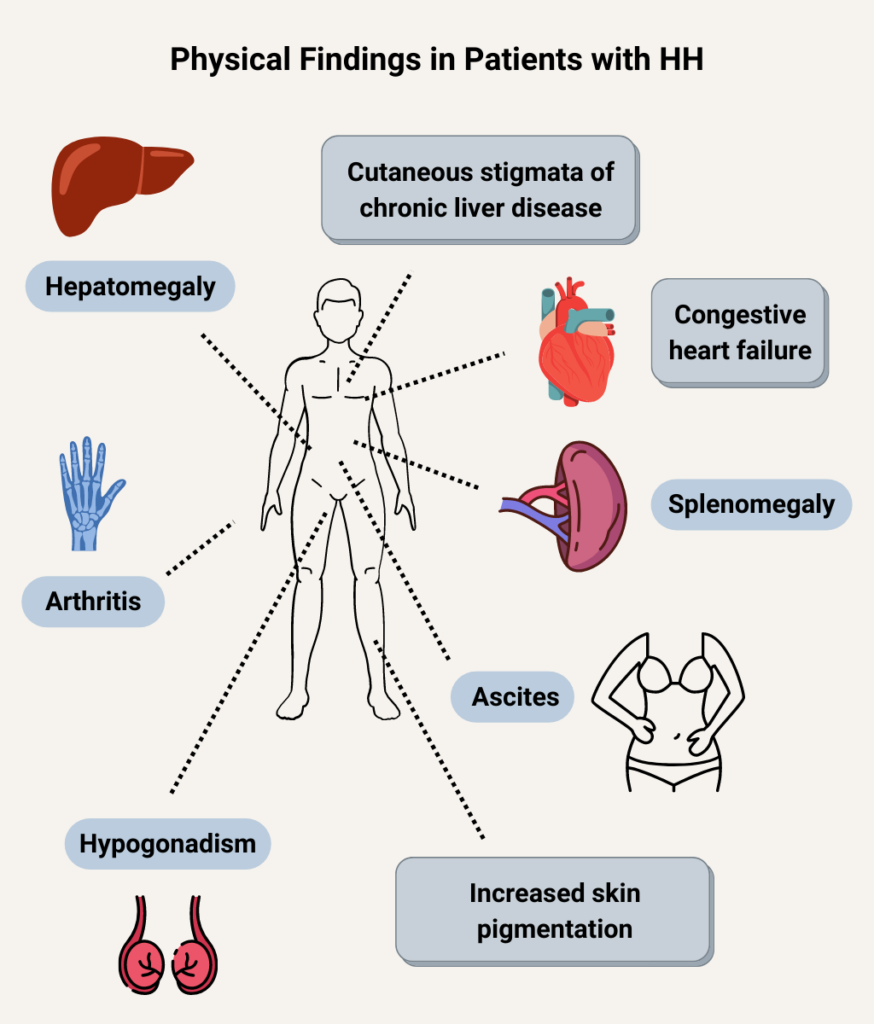
See full list of physical findings here (from ASSLD Practice Guideline).
- Asymptomatic
- Non-specific symptoms:
- Weakness
- Fatigue
- Weight loss
- Organ-related symptoms:
- Abdominal pain
- Arthralgias
- Symptoms of diabetes
- Amenorrhea
- Loss of libido, impotence
- Symptoms of heart failure, arrhythmias
C282Y and H63D mutations of the HFE gene. C282Y homozygotes account for 80%-85% of typical patients with hereditary hemochromatosis. The H63D mutation is less likely to cause disease compared to C282Y. Schematic of HFE shown here (from AASLD clinical practice guideline, PMC3149125).
- Deferoxamine (parenterally administered)
- Deferasirox (orally administered)
- Deferiprone (orally administered)
Comparison table
| Parameter | Deferoxamine | Deferasirox | Deferiprone |
|---|---|---|---|
| Route of administration | IV, SC | Oral | Oral |
| Typical dose | 30-60 mg/kg/day 5-7 days/week over 8-12 hours/day | 20-40 mg/kg once daily for dispersible tablet; 14-28 mg/kg/day for film-coated tablet or granules | 75 mg/kg/day in 3 divided doses (up to 99 mg/kg/day) |
| Half-life | 20 minutes | 8-16 hours (allows once daily dosing) | 1-3 hours |
| Major side effects | Infusion reactions, auditory and ophthalmologic effects, hypersensitivity reactions | Gastrointestinal symptoms, skin rash, transaminitis, renal dysfunction | Gastrointestinal symptoms, transaminitis, neutropenia, arthropathy |
| Pros | Available since 1970s, therefore well studied, only chelating drug acceptable in pregnancy | Oral administration, once daily | Oral administration |
| Cons | Inconvenience of parenteral administration, poor compliance | Cost | 3 divided doses (vs. once daily for deferasirox), weekly blood monitoring |
- Iron levels
- Iron increases hepcidin levels.
- Represents a feedback mechanism to maintain stable body iron levels.
- Inflammation
- Leads to increased hepcidin levels.
- Represents a host defense mechanism to limit extracellular iron availability to microbes.
- Erythropoiesis
- Increased erythroid activity leads to decreased hepcidin levels.
- Ensures iron supply for erythropoiesis.
- Erythropoietic drive overrides both iron sensing and inflammation sensing mechanisms to control hepcidin levels.
The classic presentation of advanced hereditary hemochromatosis (rarely seen today) consisting of cirrhosis, diabetes, and skin pigmentation.
Transferrin saturation. Represents the % of iron binding sites on serum transferrin that are bound by iron atoms.
A transmembrane protein expressed primarily by intestinal epithelial cells, hepatocytes and macrophages. Facilitates iron export from cells. Degraded by hepcidin. Learn more here.
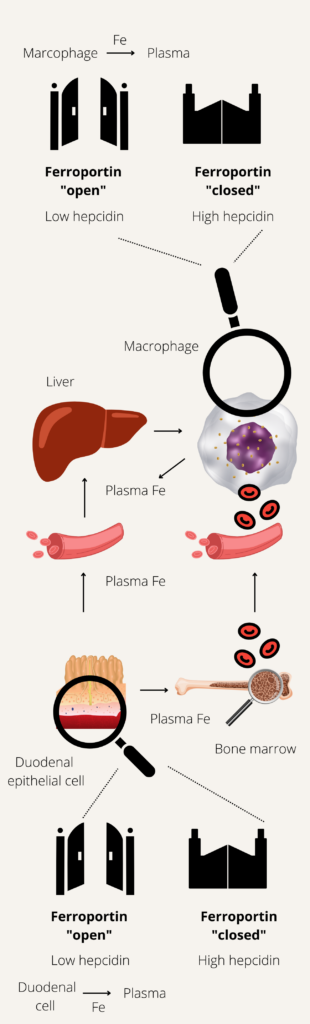
An inherited iron overload disorder associated with increased absorption of iron due to deficiency of hepcidin.
Classification of hereditary hemochromatosis:
| Classification | Genes involved | Inheritance | Protein function | Clinical manifestations |
|---|---|---|---|---|
| Type 1A HH (homozygote) | Mutations in HFE: C282Y | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 1B HH (compound heterozygote) | Mutations in HFE: C282Y H63D | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 1C HH | Mutations in HFE: S65C | AR | Possible elevations in serum iron/ ferritin, no evidence of tissue iron deposition. | |
| Type 2A juvenile HH | HJV (hemojuvelin) | AR | Involved in hepcidin synthesis, BMP co-receptor. | Earlier onset, ,30 years old, hypogonadism and cardiomyopathy are prevalent. |
| Type 2B juvenile HH | HAMP (hepcidin) | AR | Downregulation of iron efflux from erythrocytes. | Earlier onset, ,30 years old, hypogonadism and cardiomyopathy are prevalent. |
| Type 3 HH | TFR2 (transferrin receptor 2) | AR | Involved in hepcidin synthesis, interaction with transferrin. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 4A HH (FPN disease) | SLC40A1 (FPN) | AD | Duodenal iron export. | Iron deposition in the spleen is very common, lower tolerance to phlebotomies and may have anemia. |
| Type 4B HH (nonclassical FPN disease) | SLC40A1 (FPN) | AD | Resistance to hepcidin. | Fatigue, joint pain. |
Learn more here.
Measurement of iron content in liver used as reference point for estimating total body iron stores. May be quantified using:
- Liver biopsy with direct histological visualization; iron deposition:
- Is typically evaluated on a semi-quantitative scale based on Prussian Blue staining of iron granules.
- May be more accurately quantified using atomic absorption spectrophotometry.
- Magnetic resonance imaging – widely available, noninvasive procedure for estimation of LIC
- Spin-echo technique
- Gradient echo technique
Iron in the liver cannot be quantitated using ultrasound or CT scan.
Normal LIC = 0.17-1.8 mg iron/g dry weight.
Free, unbound iron in the plasma. Consists of multiple chemical structures of iron (including labile plasma iron) associated with other plasma components. Typically appear when transferrin saturation > 70%. Rapidly taken up by parenchymal cells of the liver, heart, anterior pituitary cells and pancreas, where they generate reactive oxygen species and cause organ damage.
Learn more here.
A very small fraction of total ferritin circulates in blood. In contrast to its cellular counterpart, serum ferritin does not contain any iron and is almost entirely made up of L chains. It does not play a role in iron transport or cellular iron uptake. Indeed, its function is unknown. Serum ferritin is widely used as a convenient surrogate measure of body iron stores.
About 8-9% in US.
Classification of hereditary hemochromatosis:
| Classification | Genes involved | Inheritance | Protein function | Clinical manifestations |
|---|---|---|---|---|
| Type 1A HH (homozygote) | Mutations in HFE: C282Y | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 1B HH (compound heterozygote) | Mutations in HFE: C282Y H63D | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 1C HH | Mutations in HFE: S65C | AR | Possible elevations in serum iron/ferritin, no evidence of tissue iron deposition. | |
| Type 2A juvenile HH | HJV (hemojuvelin) | AR | Involved in hepcidin synthesis, BMP co-receptor. | Earlier onset, <30 years old, hypogonadism and cardiomyopathy are prevalent. |
| Type 2B juvenile HH | HAMP (hepcidin) | AR | Downregulation of iron efflux from erythrocytes. | Earlier onset, <30 years old, hypogonadism and cardiomyopathy are prevalent. |
| Type 3 HH | TFR2 (transferrin receptor 2) | AR | Involved in hepcidin synthesis, interaction with transferrin. | Arthropathy, skin pigmentation, liver damage, diabetes mellitus, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| Type 4A HH (FPN disease) | SLC40A1 (FPN) | AD | Duodenal iron export. | Iron deposition in the spleen is very common, lower tolerance to phlebotomies and may have anemia. |
| Type 4B HH (nonclassical FPN disease) | SLC40A1 (FPN) | AD | Resistance to hepcidin. | Fatigue, joint pain. |
For a proposed new classification of HH, see Gireli et al.
Learn more here.
Phlebotomy

C282Y mutation in the HFE gene
Transferrin saturation level = (serum iron/total iron binding capacity) × 100
Increased iron intake sustained over a period of time as a result of multiple red blood cell (RBC) transfusions which may be exacerbated by increased absorption through the gastrointestinal tract.
> 45% fasting level should prompt further evaluation including HFE gene analysis.
The following recommendations are from 2019 ACG Clinical Guideline:

The following recommendations are from American Association for the Study of Liver Diseases (2011):


Raw shellfish, which may carry risk of Vibrio vulnificus (patients should also avoid iron supplements and be encouraged to reduce alcohol consumption to a minimum).
Sickle cell anemia, thalassemia major, hereditary spherocytosis, sideroblastic anemia, and pyruvate kinase deficiency.
Humans have no mechanisms to excrete iron. Iron is normally lost through:
- Skin desquamation
- Sweat – sweat contains up to 0.3 mg iron/L
- Gut
- Menstruation
- Pregnancy
Inflammation
- Primary (genetic) HLH:
- Mendelian inherited conditions leading to HLH.
- Rare in adults.
- Associated with impaired perforin-dependent cytotoxicity of T- and natural killer (NK)-cells.
- Often triggered by an infection.
- Secondary (reactive) HLH:
- Non-Mendelian HLH.
- Accounts for nearly all of adult cases.
- May occur in setting of:
- Infection – accounts for about half of HLH cases in adults.
- Malignancy – reported in about 40%-70% of adult cases.
- Autoimmune disease – also called macrophage activating syndrome [MAS] when it occurs in this patient population.
- Transplantation
Read more here.
- HLH-2004 criteria
- HScore
| Parameter | HLH-2004 diagnostic criteria | HScore | Comments |
|---|---|---|---|
| Known underlying immunosuppression | NA | 0 (no) or 18 (yes) | HIV positive or receiving long‐term immunosuppressive therapy (e.g., glucocorticoids, cyclosporine). |
| Fever | 0 (<38.5) or 1 (38.5) | 0 (<38.4), 33 (38.4–39.4), or 49 (>39.4) | Persistent or recurrent high fever. Nearly universal in untreated HLH. |
| Hepatomegaly | NA | 0 (no), 23 (hepatomegaly or splenomegaly), or 38 (hepatomegaly and splenomegaly | Abnormal liver tests reported in about 80% of adults with HLH (hypoalbuminemia detected in most adults with HLH). |
| Splenomegaly | 0 (no) or 1 (yes) | 0 (no), 23 (hepatomegaly or splenomegaly), or 38 (hepatomegaly and splenomegaly) | Splenomegaly may be caused by infiltration by activated lymphocytes and histiocytes or it by an underlying condition triggering HLH. |
| Cytopenias | 0 (one lineage) or 1 (two or three lineages)* | 0 (one lineage), 24 (two lineages), or 34 (three lineages) | Especially thrombocytopenia and anemia in early stages; results primarily from severe cytokine-mediated myelosuppression. |
| High Ferritin level (ng/ml) | 0 (<500) or 1 (500) | 0 (<2,000), 35 (2,000-6,000), or 50 (>6,000) | No specific ferritin level determined to be specific for HLH. However, the higher the ferritin the likelier the diagnosis of HLH. |
| Elevated triglyceride level | NA | 0 (<1.5), 44 (1.5-4), or 64 (>4) | Hypertriglyceridemia is detected in 42%-85% of adults. |
| Low Fibrinogen level | NA | 0 (>2.5) or 30 (<2.5) | Hypofibrinogenemia detected in about 50% of adults with HLH. |
| Elevated triglyceride level or low fibrinogen level | Triglycerides > 265 mg/dl and/or fibrinogen </= 15 mg/dL (1) | NA | |
| AST | NA | 0 (<30) or 19 (30) | LDH and bilirubin may also be increased. |
| Hemophagocytosis features on bone marrow aspirate | 0 (no) or 1 (yes) | Hemophagocytosis in bone marrow 0 (no) or 1 (yes) | If the bone marrow specimen is not conclusive, material may be obtained from other organs. |
| Elevated soluble IL-2 receptor | 1 (sIL-2R ≥ 2,400 units/mL) | NA | sIL-2R (sCD25), a marker of T cell activation; not available at most medical centers, their utility in real-time diagnosis and monitoring remains limited. |
| Low or absent NK-cell activity | 1 (Yes) | NA | Not available at most medical centers; their utility in real-time diagnosis and monitoring remains limited. |
Learn more here.
- Diagnosis of HLH
- Identification of underlying triggering condition
- Differentiating primary from secondary HLH
Learn more here.
Albumin and transferrin (or TIBC)
Ferritin, haptoglobin, fibrinogen, C-reactive protein (CRP) and hepcidin.
Alcohol is the most commonly used drug that suppresses blood cell production. Toxic effects are dose dependent. Thus, they usually occurs only in people with severe alcoholism. The following description of hematological changes in alcoholics is based on an old but excellent (and still relevant) review by Harold Ballard:
- Red cells
- Anemia :
- Sideroblastic anemia:
- Accumulation of ringed sideroblasts in the bone marrow.
- Caused by accumulation of ferritin, which can accumulate in RBC precursors, often forming granules that encircle the cell’s nucleus.
- Reduced differentiation to mature red cells, leading to anemia.
- Appear as cells with Pappenheimer bodies on peripheral smear.
- Present in 30% of severe alcoholics.
- The ringed sideroblasts generally disappear from the bone marrow within 5 to 10 days of abstinence, and RBC production resumes.
- Megaloblastic anemia – The most common cause of this deficiency is a diet poor in folic acid, a frequent complication in alcoholics, who often have poor nutritional habits.
- Spur cell hemolysis – secondary to alcoholic liver disease
- Stomatocyte hemolysis
- > 25 percent of alcoholics exhibit an increased proportion of stomatocytes in the blood.
- Stomatocytes disappear during abstinence.
- Hypophosphatemia-induced hemolysis
- Sideroblastic anemia:
- Macrocytosis (independent of folate deficiency or liver disease)
- Precise mechanism underlying macrocytosis still is unknown.
- However, alcohol appears to interfere directly with RBC development, because the macrocytes disappear within 2 to 4 months of abstinence.
- Vacuolated red cell precursors in the bone marrow:
- The vacuoles usually appear in the pronormoblasts 5 to 7 days following the initiation of heavy alcohol consumption.
- The vacuoles on average disappear after 3 to 7 days of abstinence, although in some patients they persist for up to 2 weeks
- Mechanism underlying vacuole development in blood cell precursors currently is unknown.
- Unknown whether these vacuoles affect the cell’s function and thus the drinker’s health.
- Their appearance generally is considered an indicator of excessive alcohol consumption.
- Anemia :
- Thrombocytopenia
- Leukopenia, especially neutropenia
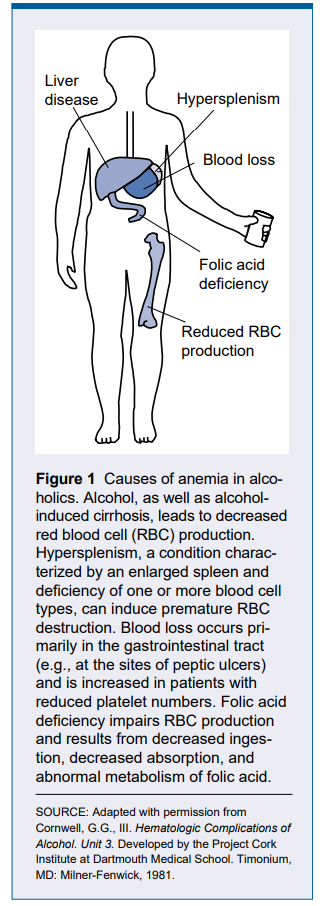
Clinical criteria for predicting the diagnosis of hemophagocytic lymphohistiocytosis (HLH):
- In 1991, the Histiocyte Society proposed a standardized set of 5 diagnostic criteria for HLH used for the prospective HLH-94 clinical trial.
- These criteria were revised for HLH-2004: individuals needed to meet 5 or more of 8 diagnostic criteria.
- Fever
- Splenomegaly
- Hypertriglyceridemia or hypofibrinogenemia
- Hemophagocytosis in bone marrow, spleen, or lymph nodes
- Low or absent NK-cell activity
- Ferritin ≥ 500 mcg/L
- Elevated soluble interleukin-2 receptor.
- The HLH-2004 criteria, developed for children, are not validated formally for adults and remain based on expert opinion.


Learn more here.
Acute intermittent porphyria
C-reactive protein
Erythrocyte sedimentation rate
Hemophagocytic lymphohistiocytosis
A macrophage that clears tissues of debris, apoptotic cells and pathogens.
Mild-moderate decrease in serum hemoglobin (Hb rarely < 8 g/dL [80 g/L]), associated with acute or chronic inflammation, in which inflammatory mediators lead to increased hepcidin expression, iron sequestration and reduced erythropoiesis. Learn more here.
C-reactive protein is a positive acute phase reactant produced by the liver. Learn more here.
Cyclosporine is an immunosuppressive that prevents T-cell activation, inhibiting interleukin-2 (IL-2) receptor expression and IL-2 production.
A transmembrane protein expressed primarily by intestinal epithelial cells, hepatocytes and macrophages. Facilitates iron export from cells. Degraded by hepcidin. Learn more here.
A condition in which body iron stores are normal or increased, but iron incorporation into red cell precursors is impaired. One example is anemia of inflammation.
HLH is an aberrant hyperinflammatory hyperferritinemic immune response syndrome that is driven by T cells and associated with a potentially fatal cytokine storm. It is characterized by fever, cytopenias, hepatosplenomegaly, and hyperferritinemia.
Phagocytosis of hematopoietic cells by activated macrophages. See image.
A 25-amino acid peptide hormone produced by liver hepatocytes in response to circulating iron levels which inhibits iron absorption from the intestinal mucosal cells and release by macrophages and hepatocytes through degradation of ferroportin-1. Hepcidin plays a critical role in iron absorption and recycling. Learn more here.
Typically refers to HLH occurring in patients with autoimmune disease and other rheumatological disorders.
Per La Rosee et al:
[MAS] refers to a subset of patients with hemophagocytic lymphohistiocytosis (HLH) arising on a background of systemic autoinflammation/autoimmunity and should be restricted to patients with Still’s disease, lupus, vasculitis, and other related autoimmune systemic diseases, because its treatment may differ from that recommended for other forms of secondary HLH.
Typically refers to HLH occurring in patients with autoimmune disease and other rheumatological disorders.
Erythrocyte sedimentation rate is the distance red cells fall by gravity in a vertical tube of anticoagulated whole blood. Learn more here.
Etoposide- and dexamethasone-based treatment protocol for patients with HLH:
- Standard of care in children.
- A more individualized approach regarding dosing (for example IV dexamethasone 10 mg/m2/day with or without modified dose of etoposide) and duration of HLH-94 is usually adopted in adults.
Consists of:
- Corticosteroids
- Cyclosporine A (CSA)
- Intrathecal therapy
- Etoposide
Goals are to:
- Delete activated T cells.
- Suppress inflammatory cytokine production.
Generates score indicating likelihood of secondary HLH based on graded clinical and laboratory parameters. HScore calculator is found here.
| Parameter | HLH-2004 diagnostic criteria | HScore | Comments |
|---|---|---|---|
| Known underlying immunosuppression | NA | 0 (no) or 18 (yes) | HIV positive or receiving long‐term immunosuppressive therapy (e.g., glucocorticoids, cyclosporine). |
| Fever | 0 (<38.5) or 1 (38.5) | 0 (<38.4), 33 (38.4–39.4), or 49 (>39.4) | Persistent or recurrent high fever. Nearly universal in untreated HLH. |
| Hepatomegaly | NA | 0 (no), 23 (hepatomegaly or splenomegaly), or 38 (hepatomegaly and splenomegaly | Abnormal liver tests reported in about 80% of adults with HLH (hypoalbuminemia detected in most adults with HLH). |
| Splenomegaly | 0 (no) or 1 (yes) | 0 (no), 23 (hepatomegaly or splenomegaly), or 38 (hepatomegaly and splenomegaly) | Splenomegaly may be caused by infiltration by activated lymphocytes and histiocytes or it by an underlying condition triggering HLH. |
| Cytopenias | 0 (one lineage) or 1 (two or three lineages)* | 0 (one lineage), 24 (two lineages), or 34 (three lineages) | Especially thrombocytopenia and anemia in early stages; results primarily from severe cytokine-mediated myelosuppression. |
| High Ferritin level (ng/ml) | 0 (<500) or 1 (500) | 0 (<2,000), 35 (2,000-6,000), or 50 (>6,000) | No specific ferritin level determined to be specific for HLH. However, the higher the ferritin the likelier the diagnosis of HLH. |
| Elevated triglyceride level | NA | 0 (<1.5), 44 (1.5-4), or 64 (>4) | Hypertriglyceridemia is detected in 42%-85% of adults. |
| Low Fibrinogen level | NA | 0 (>2.5) or 30 (<2.5) | Hypofibrinogenemia detected in about 50% of adults with HLH. |
| Elevated triglyceride level or low fibrinogen level | Triglycerides > 265 mg/dl and/or fibrinogen </= 15 mg/dL (1) | NA | |
| AST | NA | 0 (<30) or 19 (30) | LDH and bilirubin may also be increased. |
| Hemophagocytosis features on bone marrow aspirate | 0 (no) or 1 (yes) | Hemophagocytosis in bone marrow 0 (no) or 1 (yes) | If the bone marrow specimen is not conclusive, material may be obtained from other organs. |
| Elevated soluble IL-2 receptor | 1 (sIL-2R ≥ 2,400 units/mL) | NA | sIL-2R (sCD25), a marker of T cell activation; not available at most medical centers, their utility in real-time diagnosis and monitoring remains limited. |
| Low or absent NK-cell activity | 1 (Yes) | NA | Not available at most medical centers; their utility in real-time diagnosis and monitoring remains limited. |
Scoring systems used for estimating pretest probability of hemophagocytic lymphohistiocytosis (HLH):
- A Web-based online calculator developed retrospectively in adult patients that may be a helpful diagnostic tool.
- Graded clinical and laboratory parameters.

See calculator.
Caused by congenital or acquired immune dysregulation involving defects (inherited or acquired) in cytotoxic pathways of natural killer (NK) cells and/or cytotoxic T lymphocytes, leading to hypercytokinemia and an accumulation of activated lymphocytes and macrophages (termed histiocytes) in organs and tissues.
Anemia of inflammation (the anemia is not always chronic in nature).
Anemia of inflammation
Transfusion
Yes! AABB states that a single unit RBC transfusion should be standard in nonbleeding, hospitalized patients and additional units prescribed only after reassessment of patient and hemoglobin (Hb) level.
Food and Drug Administration (FDA) regulations permit hemochromatosis patients to donate blood, provided the donor (the hemochromatosis subject) meets standard blood donor eligibility criteria.
The American Red Cross does not permit hemochromatosis patients to donate blood because it “has a long-standing policy that potential donors are not allowed to receive direct compensation for their donation (beyond the usual orange juice and cookie). Because people with hemochromatosis would otherwise have to pay for their therapeutic phlebotomies, they would in effect be getting something of value for being able to donate for free. Thus the Red Cross has ruled that such donations violate their policy” (read more here).
Bottom line: blood-collection organizations can determine their own protocols within U.S. Food and Drug Administration regulations, which stipulate acceptable iron levels in donated blood.
In a 2016 Editorial, West and Eder wrote:
Safety concerns historically stem from the possibility of creating an incentive to donate blood for free rather than to pay for therapeutic phlebotomy, possibly encouraging HH donors to deny risk factors for infectious diseases. In the Final Rule that became effective in May 2016, the US Food and Drug Administration codified the requirements for hereditary hemochromatosis (HH) donors in the Code of Federal Regulations (CFR), thus eliminating the need for a variance to collect whole blood more frequently than every 8 weeks (or double red blood cells more frequently than every 16 weeks) and distribute units without special labeling from HH donors who meet all eligibility requirements. Notably, the CFR retains the requirement for obtaining a prescription for therapeutic phlebotomy from a licensed health care provider and performing therapeutic phlebotomy free of charge… Since the molecular basis of the disease was elucidated in 1996, it has been posited that the condition itself poses no harm to the recipient. The question concerns the motives of the HH donor to give blood and the possibility of incentive to withhold information from blood establishments about infectious risk factors
In 2016, the American Red Cross wrote (despite its continued refusal to allow patients with HH to donate blood) a piece titled “Iron-rich blood is just fine, thank you!”:
For decades, blood centers in the United States would not collect whole blood from donors/patients with hereditary hemochromatosis (HH), in some cases because it used to be that such units had to be labeled with the disease necessitating its removal… In 2016, the FDA encoded the regulations for therapeutic phlebotomy… Special labeling is not required, and units may be distributed if they meet regular requirements and criteria, as long as the therapeutic phlebotomy (TP) is ordered by a physician and the phlebotomy performed without charge.
CCI = posttransfusion count increment (CI; posttransfusion platelet count – pretransfusion platelet count) × body surface area (in m2)/number of platelets transfused
See calculator.
Posttransfusion platelet count – pretransfusion platelet count
By apheresis or from whole blood by platelet-rich plasma or buffy coat methods.
They are stored in gas-permeable storage bags with gentle agitation at 20-24 degrees C (68-75.2 degrees F).
Removes autoantibodies and replenishes ADAMTS13 levels.
Leukocyte reduction filters are used to remove > 99.9% of leukocytes. According to a Circular of Information from the AABB, American Red Cross, America’s Blood Centers, and the Armed Services Blood Program, leukocyte-reduced units of red blood cells must have a residual content of leukocytes <5.0 x 106.
FDA-accepted minimum levels of Hb:
- For men – ≥ 13 g/dL (130 g/L) or Hct level ≥ 39%
- For women – ≥ 12.5 g/dL (125 g/L) or Hct level ≥ 38%
Per Technical Manual, 20th Edition:

Massive transfusion refers to the transfusion of a large volume of blood products within a short period of time and is typically defined as transfusion of ≥ 10 red blood cell (RBC) units within 24 hours. Read more here.
2019 Guidelines on the Use of Therapeutic Apheresis in Clinical Practice:
[Plasma exchange] is generally performed daily until the platelet count is >150 × 109/L, and LDH is near normal for 2-3 consecutive days. The role of tapering therapeutic plasma exchange (TPE) over longer duration has not previously been studied prospectively but is currently being reviewed. A small retrospective study suggests a lower overall recurrence rate at 6 months with taper. A common taper strategy is three times a week for the first week, twice weekly the second and then once weekly the following week(s). Other taper approaches have been documented.
From whole blood or by apheresis
≤ 5 days in the United States; however, most blood collection centers culture apheresis platelets and release the unit 24-36 hours after collection.
Up to 42 days.
- 21 days if stored in citrate phosphate dextrose (CPD), or CPD2 anticoagulant-preservative
- 35 days if stored in citrate phosphate dextrose adenine 1 (CPDA-1)
- 42 days using current generation of additive solutions additive solutions
1 apheresis platelet unit is equivalent to a pool of 4-6 units of whole blood-derived platelets; typically contains ≥ 3 × 1011 platelets.
100 million per year worldwide, 13 million in the US.
Each unit of RBCs (processed from 420 mL of donor blood) contains about 200 mg of iron (0.47 mg iron/mL of whole donor blood or 1.08 mg iron/mL of pure RBCs).
Yes, typically for complement only. However, it is also weakly positive for IgG in about one quarter of cases. Learn more here.
TRALI risk per component reported to be 7 times higher for plasma than for red blood cells.
- Reduce rates of alloimmunization.
- Reduce rates of febrile nonhemolytic transfusion.
- Prevent transmission of intracellular pathogens such as:
- Cytomegalovirus (CMV)
- Human T-lymphotropic virus
- Types I and II (HTLV- I/II)
- Ratio-driven transfusion – a fixed ratio of red cells, platelets and plasma components is administered.
- Goal-directed transfusion – lab test results are used to guide decision about transfusion of blood components.
Learn more here.
- Acute hemolytic transfusion reaction (AHTR)
- Febrile nonhemolytic transfusion reactions (FNHTR)
- Urticaria
- Anaphylaxis
- Transfusion-related acute lung injury (TRALI)
- Transfusion-associated circulatory overload (TACO)
- Nonimmune hemolysis
- Hypotensive transfusion reactions
- Transfusion-associated sepsis
- Immune-mediated:
- Alloimmunization to human leukocyte, platelet-specific, or ABO antigens
- Autoimmune (immune thrombocytopenia)
- Non-immune-mediated:
- Hemorrhage
- Fever
- Sepsis
- Disseminated intravascular coagulation (DIC)
- Splenic sequestration
- Use of older stored platelets
- Thrombotic microangiopathy
Learn more here.
- Transfusion-associated graft-versus-host disease
- Immunocompromised patients
- Hematologic malignancies or certain solid tumors
- Recipients of marrow or peripheral blood stem cell transplantation
- Patients receiving blood components from blood relatives or human leukocyte antigen-compatible donors
- Patients receiving fludarabine therapy
- Prematurity, low birthweight, or erythroblastosis fetalis in newborn
- Those at risk for transfusion-associated graft-versus-host disease (TA-GVHD) caused by proliferation of donor T lymphocytes
- Those who are immunocompromised
- Those receiving intrauterine transfusion
- Those with hematologic malignancies or solid tumors, including:
- Sarcoma
- Neuroblastoma
- Hodgkin lymphoma
- Those who are recipients of marrow or peripheral blood stem cell transplantation
- Those receiving RBCs from blood relatives or human leukocyte antigen-compatible donors
- Those receiving fludarabine therapy
- Those receiving granulocyte transfusions
According to the AABB:
Description:
- Blood components that contain viable lymphocytes may be irradiated to prevent proliferation of T lymphocytes, which is the immediate cause of TA-GVHD.
- Irradiated blood is prepared by exposing the component to a radiation source.
- The standard dose of gamma or X-ray irradiation is 2500 centigray (cGy) targeted to the central portion of the container with a minimum dose of 1500 cGy delivered to any part of the component.
Indications:
- Patients at risk for TAGVHD, including:
- Fetal and neonatal recipients of intrauterine transfusions
- Selected immunocompromised recipients
- Recipients of cellular components known to be from a blood relative
- Recipients who have undergone peripheral blood progenitor cell transplantation
- Recipients of cellular components whose donor is selected for HLA compatibility and recipients of granulocyte transfusions.
- Patients receiving purine analogues (eg, fludarabine, cladribine) or certain other biological immunomodulators (eg, alemtuzumab, antithymocyte globulin) who may be at risk for TA-GVHD, depending on clinical factors and the source of the biological agent.
Patients with low circulating platelet counts or functionally abnormal platelets, to prevent or control bleeding.
- Patients who are IgA deficient with documented presence of antibodies against IgA, for whom no IgA-deficient products are available.
- Patients experiencing posttransfusion purpura (washing can help remove complement).
- Patients with history of repeated severe allergic reactions to plasma-containing products.
- Patients with history of severe allergic reactions to plasma-containing products.
- Patients who have absolute immunoglobulin A (IgA) deficiency and for whom no IgA-deficient RBCs are available.
- Patients at risk of hyperkalemia.
- Neonates with neonatal alloimmune thrombocytopenia requiring maternal RBC transfusion that contains antihuman platelet antigen-1a (however, use of washed RBCs is not required.)
According to AABB:
- Description:
- Washed components are typically prepared using 0.9% Sodium Chloride, Injection USP with or without small amounts of dextrose.
- Washing removes unwanted plasma proteins, including antibodies and glycerol from previously frozen units.
- The shelf life of washed components is no more than 24 hours at 1 to 6 C or 4 hours at 20 to 24 C.
- Washing is not a substitute for leukocyte reduction, and only cellular components should be washed.
- Indications:
- To reduce exposure to antibodies targeting known recipient antigens.
- To remove constituents that predispose patients to significant or repeated transfusion reactions (eg, removal of IgA-containing plasma in providing transfusion support for an IgA-deficient recipient or in rare recipients experiencing anaphylactoid/anaphylactic reactions to other plasma components).
Thrombotic thrombocytopenic purpura (TTP) and heparin-induced thrombocytopenia (HIT)
Platelets collected using automated instrumentation (also called apheresis platelets).
- Reduce rates of alloimmunization
- Reduce rates of febrile nonhemolytic transfusion reactions
- Prevent cytomegalovirus transmission
Urticaria and febrile nonhemolytic transfusion reactions (FNHTRs)
- Fresh frozen plasma (FFP) – In freezer (at < -18C) within 8 hours of collection.
- Plasma Frozen Within 24 Hours After Phlebotomy (PF24):
- In refrigerator (at 1-6C) within 8 hours of collection.
- In freezer (at < -18C) within 24 hours of collection.
- Plasma Frozen Within 24 Hours After Phlebotomy Held At Room Temperature Up To 24 Hours After Phlebotomy (PF24RT24):
- Room temperature (20-24C) for up to 24 hours after collection.
- In freezer (at < -18C) within 24 hours of collection.
Normal levels of coagulation factors, antithrombin, and ADAMTS13.
Packed red blood cells, used for transfusion.
Transfusion-associated circulatory overload
Transfusion-related acute lung injury
Based on a landmark randomized study in 1991 (see abstract below), plasma exchange was reported to be associated with a mortality rate of about 10%, vs. 90% in historical controls.

FDA test requirements (with American Red Cross guidance):
- Hepatitis B virus (HBV):
- The tests used for blood donor screening are the GS HBsAg EIA 3.0, a qualitative ELISA for the detection of Hepatitis B Surface Antigen (HBsAg), and the Ortho HBc ELISA for the qualitative detection of antibody to HBV core antigen (anti-HBc) in human serum and plasma samples.
- An FDA licensed triplex nucleic acid test (NAT) using transcription-mediated amplification was introduced by the Red Cross in 2009. The assay detects HBV DNA, HIV-1 RNA, and HCV RNA.
- The frequency of detecting an active HBV infection in a blood donor is about 1 per 12,000 donations screened.
- The per-unit risk of HBV infection through blood transfusion is less than 1 per million units screened.
- Hepatitis C (HCV):
- The test used for blood donor screening is the Ortho HCV ELISA for the qualitative detection of antibody to HCV antibodies (anti-HCV) in human serum or plasma samples.
- A duplex nucleic acid test (NAT) was introduced for HIV-1/HCV RNA detection in 1999 and updated to include the detection of HBV DNA in 2009.
- Donors who test HCV-antibody reactive, but NAT nonreactive by routine testing are further tested individually for HCV RNA by NAT.
- Donors who test anti-HCV and HCV NAT reactive do not require further testing.
- The frequency of detecting a positive donor is about 1 per 5,000 donations screened.
- The per-unit risk of HCV infection through blood transfusion is less than 1 per 2 million units screened.
- HIV:
- The test used for blood donor screening is the GS HIV-1/HIV-2 PLUS O EIA for the simultaneous qualitative detection of anti-HIV 1 (groups M and O) and/or HIV-2 in human serum or plasma.
- A duplex nucleic acid test (NAT) was introduced for HIV-1/HCV RNA detection in 1999 and updated to include the detection of HBV DNA in 2009.
- Donors who test antibody reactive are further evaluated by additional tests to confirm the presence of HIV antibody and to differentiate HIV-1 from HIV-2 antibodies.
- Donors who test anti-HIV-1/HIV-2 and HIV-1 NAT reactive are not further tested.
- The frequency of detecting HIV-1 in a blood donor is about 1 per 33,000 donations screened.
- detecting HIV-2 in a blood donor is extremely rare at 1 per 57 million donations, with only 5 such infected donors ever identified since HIV-2 screening began in 1992.
- The per-unit risk of HIV-1 infection through blood transfusion is less than 1 per 2 million units screened.
- Human T-cell lymphotropic virus I/II (HTLV-I/II):
- The test used for blood donor screening is the Avioq HTLV-1/2 Microelisa system for the qualitative detection of antibodies to HTLV-1 and HTLV-2 in human serum or plasma samples.
- Donors who test reactive for anti-HTLV-1/2 are further tested using an FDA licensed western blot to determine if antibodies are present.
- There are no nucleic acid tests (NAT) available for HTLV-1/2.
- The per-unit risk of transfusion-transmitted HTLV-1/2 is less than 1 per 2 million units screened, and the frequency of detecting an infected donor is 1 per 27,000 donations screened.
- Syphilis:
- Screening for syphilis is performed using a qualitative test that detects the presence of antibodies to the spirochete (corkscrew-shaped bacterium), Treponema pallidum, by an automated agglutination assay based on specific pattern recognition. C
- Confirmation is performed using another serologic test for total antibodies, an enzyme-linked immunoassay, as well as a test for reagin (a protein-like substance that is present during acute infection and for several months following resolution of infection).
- No cases of transfusion-transmitted syphilis have been recorded in more than 50 years.
- West Nile virus (WNV):
- WNV RNA is detected by an FDA licensed NAT assay similar to that used for HIV-1, HCV, HBV, and Zika virus.
- NAT-reactive donations are further tested by repeat NAT and antibody to confirm infection.
- Following the introduction of blood donor screening, there have been 15 cases of transfusion transmission from screened blood; all are believed to be due to donations having very low levels of virus. This translates to a risk of about 1 per 84 million donations for the Red Cross overall (or 1 per 35 million during the summer transmission season).
- Trypanosoma cruzi:
- The Red Cross blood donations are screened using the Ortho T. cruzi Enzyme-Linked Immunosorbent Assay (ELISA) for the qualitative detection of antibodies to T. cruzi in human serum or plasma samples.
- An FDA licensed enzyme strip immunoassay (ESA) is used for confirmatory testing.
- Because T. cruzi is not endemic in the United States, the Red Cross (and all US blood centers) donors are tested only once.
- The frequency of detecting a positive donor is about 1 per 15,000 first-time donations screened.
- Babesiosis:
- In May 2018, the Red Cross began testing from whole blood samples using a NAT assay that detects the main four species of babesia pathogenic to humans. The assay, now FDA licensed, detects ribosomal RNA of the parasite following red cell lysis, significantly increasing sensitivity and obviating the need for antibody testing.
- Tested in 14 states in the US where incidence of infection is highest.
Refers to changes in quality and quantity of packed red blood cells (pRBCs) during storage, including:
- Change in cell shape change and microvesiculation
- Decrease in adenosine triphosphate (ATP) and 2,3-diphosphoglycerate levels
- Increase in lysophospholipids
- Increase in potassium
- Increase in free hemoglobin (Hb)
The storage lesion represents a risk to efficient RBC perfusion and tissue oxygen delivery, and it has been suggested that it may result in adverse outcomes. This has led to the classification of “young” (<14-21 days) vs “old” (>21 days) RBC units. According to the AABB, 13 randomized control trials have evaluated the effect of RBC storage duration of transfused RBCs on patient outcomes (7 since 2012). However, there is currently no formal guidance on the optimal length of RBC storage prior to transfusion. The AABB recommends administering RBC units within licensed expiration date (standard issue) rather than transfusing only fresh RBCs (< 10 days old) to patients, including neonates.
Serological or electronic crossmatches are performed to detect serological incompatibility (the presence of antibodies) between recipient and donor before transfusion.
According to the AABB:
DAT (also called a Coombs test) detects immunoglobulins and complement bound to red blood cells.
Defined as ≥ 1 degrees C increase in temperature ≥ 38 degrees C (100.4 degrees F) that is associated with transfusion and no other causes are evident. Caused by antibody to donor leukocytes. Symptoms include:
- Fever
- Chills
- Tachypnea
- Headache
- Vomiting
According to the AABB, febrile nonhemolytic transfusion reaction defined as fever and/or chills without hemolysis occurring
in the patient during or within 4 hours of cessation of transfusion. If transfusion-related, the most common cause is a reaction to passively transfused cytokines or a reaction of recipient antibodies and leukocytes in the blood product. If blood culture of patient or residual component is performed, the results should be negative. Laboratory findings should show no evidence of
acute hemolysis.
Treatment includes antipyretic such as acetaminophen or meperidine (for more severe reactions).
Administer leukocyte-reduced blood or washed RBCs to reduce risk.
A platelet transfusion to prevent bleeding.
A transfusion (e.g. of fresh frozen plasma or platelets) to prevent bleeding.
IV transfusion without removing patient blood volume.
A transfusion (e.g. of fresh frozen plasma or platelets) to treat bleeding.
An adverse event that occurs during or after transfusion and that is caused by transfusion of that blood component.
Testing for presence of A and B antigens on red blood cells:
- Forward typing – red blood cells (RBCs) are tested for A and B antigens.
- Reverse typing – serum or plasma is screened for presence of anti-A and anti-B antibodies.
According to a Circular prepared jointly by AABB, the American Red Cross, America’s Blood Centers, and the Armed Services Blood Program:
The ABO group of all red-cell-containing components must be compatible with ABO antibodies in the recipient’s plasma. Serologic compatibility between recipient and donor must be established before any red-cell containing component is transfused. This may be accomplished by performing ABO/Rh typing, antibody screening, and crossmatching by serologic technique or use of a computer crossmatch.
Acute, severe reaction, typically caused by red blood cell ABO incompatibility.
According to the AABB, acute hemolytic transfusion reaction is defined as the rapid destruction of red blood cells during,
immediately after, or within 24 hours of cessation of transfusion. Clinical and laboratory signs of hemolysis are present.
Criteria for diagnosis:
According to the AABB, an allergic reaction is the the result of an interaction of an allergen with preformed antibodies. In some instances, infusion of antibodies from an atopic donor may also be involved. It may present with only muccocutaneous signs and symptoms.
Criteria for diagnosis:
ABO compatibility verified using computer system without immediate spin or and antiglobulin crossmatching if significant alloantibodies have not been detected in the recipient sample.
Using AABB guidelines, the electronic crossmatch is possible only if the following conditions are met:
- The patient’s ABO group and Rh type has been done twice and entered in the computer (one group can consist of a record but one must be done on a current in-date specimen). * The computer must alert the technologist if there is a discrepancy between the two groups.
- The donor ABO (and Rh types, if negative) have been confirmed and entered in the computer. The donor’s unit identification number, component name, and ABO/Rh type must also be entered in the computer (manually or by scanning the bar code label on the unit).
- The computer system will alert the technologist to ABO & Rh discrepancies between information on the donor label and results of donor confirmatory testing.
- The computer system will alert the technologist to ensure correct data entry and interpretation, e.g., prevent group O test results from being misinterpreted as group A.
- The computer system will alert the technologist to ABO and Rh discrepancies patient and donor groups. The program should be programed to prevent assigning ABO incompatible blood (e.g., group A red cells to a group O recipient) and to give an alert when assigning Rh-positive red cells to Rh-negative recipients.
A byproduct of cryoprecipitate production generated after the removal of cryoprecipitate from thawed fresh frozen plasma centrifuged at 1-6 degrees C.
A blood product made by thawing fresh frozen plasma at 1-6 degrees C and recovering the cold insoluble precipitate by centrifugation. The precipitate is resuspended in a small quantity of plasma and refrozen/stored at at -18 to -25 degrees C.
Collection:
Fresh frozen plasma (FFP) is plasma that is prepared from a whole blood or apheresis collection and frozen at –18 C or colder within the time frame as specified in the manufacturer’s instructions for use of the blood collection, processing, and storage system (typically within 8 hours of phlebotomy):
- On average, units contain 200 to 250 mL, but apheresis-derived units may contain as much as 400 to 600 mL. FFP contains plasma proteins, including all coagulation factors.
- FFP contains normal levels of the labile coagulation factors, factors V and VIII.
- FFP should be infused immediately after thawing or stored at 1 to 6 C. After 24 hours, the component must be discarded or, if collected in a functionally closed system, may be relabeled as thawed plasma.
Recipient-donor compatibility:
Plasma must be ABO compatible with the recipient’s red cells. Compatibility with RhD is not necessary in plasma transfusion. Compatibility tests prior to transfusion are not necessary.
Indications:
FFP serves as a source of plasma proteins for patients who are deficient in or have defective plasma proteins.
Read more here.
Measurement of iron content in liver used as reference point for estimating total body iron stores. May be quantified using:
- Liver biopsy with direct histological visualization; iron deposition:
- Is typically evaluated on a semi-quantitative scale based on Prussian Blue staining of iron granules.
- May be more accurately quantified using atomic absorption spectrophotometry.
- Magnetic resonance imaging – widely available, noninvasive procedure for estimation of LIC
- Spin-echo technique
- Gradient echo technique
Iron in the liver cannot be quantitated using ultrasound or CT scan.
Normal LIC = 0.17-1.8 mg iron/g dry weight.
Pathogen-reduced plasma is plasma that has been subjected to reduction technologies such as methylene blue, amotosalen or riboflavin treatment plus ultraviolet light, and solvent/detergent treatment to minimize transfusion-associated infections.
Pathogen reduced plasma products: a clinical practice scientific review from the AABB concluded in 2019:
The available evidence indicates that PR plasma products are as efficacious as conventional plasma products in a variety of patient populations and clinical conditions, except in the case of methylene blue and riboflavin PI…. With regard to non-infectious AE, this review has summarized evidence which, in aggregate, indicates that currently available S/D and PR plasma products have no documented deleterious effects compared to conventional plasma products. Furthermore, they appear to have lower rates of allergic reactions and TRALI.
Learn more here.
A fresh frozen plasma product in which the sample is placed:
- In refrigerator (at 1-6C) within 8 hours of collection.
- In freezer (at < -18C) within 24 hours of collection.
Plasma frozen within 24 hours (PF24) of blood collection.
PF24RT24 stands for “Plasma frozen within 24 hours after phlebotomy held at room temperature for up to 24 hours”:
- Room temperature (20-24C) for up to 24 hours after collection.
- In freezer (at < -18C) within 24 hours of collection.
According to the AABB, plasma is the aqueous part of blood containing proteins and salt in which red and white blood cells and platelets are suspended. It constitutes approximately 55 percent of total blood volume. Important elements in plasma include albumin, coagulation factors, fibrinolytic proteins, immunoglobulin and other proteins.
Repeated failure to achieve adequate response to platelet transfusions
Genetic or acquired conditions associated with absent or reduced numbers of platelet granules or granules that are unable to empty their contents into the bloodstream. Learn more here.
FFP, PF24, or PF24RT24 thawed and stored up to 5 days or original date of expiration (whichever is sooner) at 1-6 degrees C (33.8-42.8 degrees F).
Screening for complement-activating alloantibodies in a patient who will receive a blood transfusion, using commercially available group O RBCs that express all antigens as required by the FDA.
Per the AABB:
Pretransfusion compatibility testing begins with the type and screen procedure. The recipient’s ABO group and Rh type are determined first; then a screening procedure is used to detect any unexpected non-ABO blood group antibodies that may be present. If the screening test reveals the presence of an antibody, the specificity of that antibody is determined by an antibody identification panel. Once the specificity of the antibody has been identified, donor units of the appropriate ABO group and Rh type are screened for the corresponding antigen. Units that are negative for that antigen are crossmatched with the recipient to ensure compatibility.
Each unit of RBCs contains enough hemoglobin to increase the hemoglobin concentration in an average-sized adult by approximately 1 gram/deciliter (g/dL) (increase hematocrit by 3%). Learn more here.
In one study, 1/1,700 units.
AB blood (about 4%).
O blood (45%-50%).
What is the most common cause of transfusion-related morbidity and mortality in the United States?
Transfusion-associated circulatory overload (TACO)
15%-17% in persons of European ethnicity, 8% in persons with African ethnicity, < 0.1% in persons of Asian ethnicity.
RBC transfusion at Hb level of < 7 g/dL (70 g/L) with a target Hb of 7-9 g/dL (70-90 g/L). Transfusion trigger should not exceed 90 g/L in most critically ill patients.
AABB recommendations in 2016:
Consider treatment of symptomatic anemia with red cell transfusion to minimize symptoms; transfusion usually required when Hb is < 6 g/dL (60 g/L).
For patients having orthopedic or cardiac surgery and are hospitalized and hemodynamically stable, transfuse at Hb level ≤ 8 g/dL (80 g/L).
AABB recommendations in 2016:
> 24 hours after transfusion.
Within 24 hours of transfusion or during transfusion.
300 mL to 400 mL
According to a Circular prepared jointly by AABB, the American Red Cross, America’s Blood Centers, and the Armed Services Blood Program:
Depending upon the collection system used, a single whole blood donation typically contains either 450 mL (+/- 10%) or 500 mL (+/- 10%) of blood collected from allogeneic blood donors. After plasma is removed, the resulting component is RBCs, which has a hematocrit between 65% to 80% and a usual volume between 225 mL and 350 mL. Red Blood Cells additive solutions (AS) may be mixed with the red cells remaining after removal of nearly all of the plasma to extend the shelf life (see Table 2). The typical hematocrit of AS RBCs is 55% to 65%, and the volume is approximately 300 to 400 mL.
Acute pulmonary edema due to volume overload from transfusion. Usual onset within 1-2 hours of transfusion. Risk factors include transfusion of large volumes of components or high flow rates. Treatment is supportive and may include lasix, and rarely therapeutic phlebotomy.
According to Nareg Roubinian, TACO is pulmonary edema primarily related to circulatory overload with criteria developed by the National Healthcare Safety Network including 3 or more of the following within 6 hours of transfusion:
- Acute respiratory distress
- Radiographic pulmonary edema
- Elevated central venous pressure
- Evidence of left heart failure
- Elevated B-type natriuretic peptide (BNP)
- Positive fluid balance
According to the AABB, TACO is defined as new onset or exacerbation of 3 or more of the following within 12 hours of cessation of transfusion:
- Elevated brain natriuretic peptide (BNP) or NT-pro BNP relevant biomarker.
- Evidence of cardiovascular system changes not explained by underlying medical condition (Elevated central venous pressure, evidence of left heart failure including development of tachycardia, hypertension, widened pulse pressure, jugular venous distension, enlarged cardiac silhouette and/or peripheral edema).
- Evidence of fluid overload.
- At least 1 of the following:
- Evidence of acute or worsening respiratory distress (dyspnea, tachypnoea, cyanosis and decreased oxygen saturation values in the absence of other specific causes) and/or,
- Radiographic or clinical evidence of acute or worsening pulmonary edema (crackles on lung auscultation, orthopnea, cough, a third heart sound and pinkish frothy sputum in severe cases).
Form of acute lung injury caused by donor leukocyte antibodies (occasionally in recipient) and other leukocyte activating agents in plasma-containing components including whole blood, RBCs, platelets, cryoprecipitate, and fresh frozen plasma. Symptoms arise within 6 hours of transfusion and typically resolve after 48-96 hours. Treatment is supportive.
The Canadian Consensus Criteria defines TRALI as acute pulmonary edema after transfusion in the absence of circulatory overload or alternate acute respiratory distress syndrome (ARDS) risk factors:
According to the AABB, TRALI is defined as:
- No evidence of acute lung injury (ALI) prior to transfusion, and
- ALI onset during or within 6 hours of cessation of transfusion, and
- Radiographic evidence of bilateral infiltrates, and
- No evidence of left atrial hypertension (i.e., circulatory overload), and
- Hypoxemia defined by any of these methods:
- PaO2/FiO2 less than or equal to 300 mmHg
- Oxygen saturation less than 90% on room air
- Other clinical evidence
Increased iron intake sustained over a period of time as a result of multiple red blood cell (RBC) transfusions which may be exacerbated by increased absorption through the gastrointestinal tract.
About 25%-35%. Learn more here.
Immunocompromised patients, those at risk for transfusion-associated graft vs. host disease, patients with cancer, recipients of bone marrow transplantation, patients receiving fludarabine.
Coagulation
Yes. Desmopressin/DDAVP (1-deamino-8-D-arginine vasopressin) is a synthetic analogue of hormone 8-arginine vasopressin, an antidiuretic hormone.
Yes, they are 20-25% lower in individuals with type O blood group. 14% of individuals with type O blood have vWF levels ≤ 50 units/dL. Learn more here.
No. In unconfirmed aHUS, serum/plasma complement factor levels may suggest the diagnosis but they are not diagnostic.
Fragmented RBCs can be detected by certain automated hematology analyzers based on the analysis of fraction of small red blood cells (RBCs) in the context of normal RBC volume indices (mean corpuscular volume and width). Their presence should prompt a peripheral blood smear review.
Yes, DVT may rarely occur in arms of the upper extremity, splanchnic veins and cerebral veins.
No, they should be avoided in pregnancy. Vitamin K antagonists are also contraindicated in pregnancy. Pregnant women requiring anticoagulation should receive low molecular weight heparin.

Yes, and in fact they are now recommended as an option in the latest American Society of Hematology (ASH) guideline, especially for those who are not at high risk of bleeding and who do not have extensive clot burden:

No, factor XIII deficiency cannot be detected by routine clotting tests such as PT, aPTT, or thrombin time. Rather, it is detected by ammonia release or amine incorporation assays.
There is an increasing trend towards using DOACs in this clinical context based on recent clinical trials. For example, it was shown that edoxaban (a DOAC used in Europe) may be as effective as low-molecular-weight heparin (LMWH) in reducing venous thromboembolism (VTE) recurrence/bleeding. The latest guidelines from ACCP and NCCN (see below) recommend administering oral factor Xa inhibitor (apixaban, edoxaban, or rivaroxaban) rather than LMWH during initiation and treatment phase of deep vein thrombosis (DVT). Because edoxaban and rivaroxaban reported to be associated with higher risk of gastrointestinal major bleeding compared to LMWH in patients with luminal gastrointestinal malignancy, apixaban or LWMH may be preferred in these patients. Learn more here.



At minimum, all critically ill non-bleeding patients with DIC should receive prophylactic anticoagulation for venous thromboembolism.
Regarding therapeutic doses of heparin, the 2013 ISTH harmonization guideline states:
Anticoagulant treatment may be a rational approach based on the notion that DIC is characterized by extensive activation of coagulation. Although experimental studies have shown that heparin can at least partly inhibit the activation of coagulation in DIC, there are no RCTs demonstrating that the use of heparin in patients with DIC results in an improvement in clinically relevant outcomes… therapeutic doses of heparin should be considered in cases of DIC where thrombosis predominates (low quality evidence). The use of low molecular weight heparin (LMWH) is preferred to the use of unfractionated heparin (UFH) in these cases (low quality evidence).
Patients with type 2B vWD may have thrombocytopenia. Learn more here.
In patients with synthetic liver dysfunction, the vitamin K cycle is impaired, so even large doses of vitamin K are not effective. Partial response may be seen in those patients who are vitamin K deficient, for example those receiving broad-spectrum antibiotics. Example of a negative study here.
Italian Association for the Study of Liver Diseases (AISF) and the Italian Society of Internal Medicine (SIMI) (AISF/SIMI) recommends against the routine administration of vitamin K to increase the plasma levels of coagulation factors in patients with cirrhosis:

IIV and oral vitamin K result in similar INR reversal at 24-72 hours, but onset of action is more rapid with IV vitamin K (4-6 hours) compared with oral (18-24 hours).
All are equally efficacious in patients without cancer and with normal renal function.


No, because neither test accurately reflects the in vivo hemostatic balance. 2016 AASLD practice guideline: “INR is not a reliable indicator of coagulation status in cirrhosis”.
One daily dose is sufficient. Recommended s.c. adult dosing of enoxaparin (Lovenox): 1 mg/kg twice daily or 1.5 mg/kg/day. Learn more here.
Generally no, unless the patient has underlying kidney disease or is pregnant.
Because these patients have reduced levels of both procoagulants and anticoagulants, and the INR/PT and aPTT do not detect changes in anticoagulants.
About 6.5 adverse events per 10,000 units reported in Canada. Such reactions include allergic reactions, transfusion-related acute lung injury (TRALI) and transfusion-associated circulatory overload (TACO). Learn more here.
15%-20% of patients presenting with acute lymphoblastic leukemia, > 90% of patients presenting with acute promyelocytic leukemia.
Distal DVT 20%, proximal DVT 80%
Lower rates of pulmonary embolism, DVT recurrence and postthrombotic syndrome.
Comparable protection against venous thromboembolism (VTE) recurrence, less overall risk of bleeding (especially intracranial bleeding). Studies have shown that gastrointestinal bleeding may be increased in patients taking DOAC but these studies involve cohorts with atrial fibrillation, not VTE.
Discontinue warfarin and start dabigatran or apixaban when INR <2.0
Learn more here.
Discontinue warfarin and start rivaroxaban when INR <3.0.
Learn more here.
DDAVP 0.3 mcg/kg IV in 30-50 mL normal saline over 30 minutes. May be repeated 12-24 hours after first dose, depending on how well the patient responds, but no more than 2-3 doses should be given in order to avoid tachyphylaxis.
- No FDA-approved reversal agents for fondaparinux; no specific antidote available.
- Fondaparinux does not bind to protamine sulfate; therefore protamine sulfate is not recommended.
- Consider activated PCC (aPCC) 20 units/kg.
- If aPCC is contraindicated or not available, consider rFVIIa 90 mcg/kg.
For IV heparin:
- Protamine sulfate dosing is 1 mg per 100 units of heparin, if heparin was given in the previous 2-3 hours.
- Protamine sulfate is given slower than 5 mg/minute to minimize the risk of adverse reactions.
- Maximum dose of protamine sulfate is 50 mg.
For SC heparin:
- When heparin has been given subcutaneously (and therefore is cleared with a longer half-life) prolonged or repeated administration of protamine sulfate may be required.
- Dabigatran:
- Overlap warfarin with dabigatran for 3 days (normal renal function); 2 days (CrCl 30 to 50 mL/min); or 1 day (CrCl 15 to 30 mL/min); note that dabigatran can contribute to INR elevation or,
- Overlap warfarin with dabigatran until the INR is therapeutic on warfarin.
- Apixaban:
- If continuous anticoagulation is needed, discontinue apixaban and start a parenteral anticoagulant with warfarin; continue the parenteral agent until the INR is therapeutic on warfarin. Note that apixaban can contribute to INR elevation or,
- Overlap warfarin with apixaban until the INR is therapeutic on warfarin, testing right before the next apixaban dose to minimize the effect of apixaban on INR elevation.
- Rivaroxaban:
- Discontinue rivaroxaban and start a parenteral anticoagulant with warfarin; continue the parenteral agent until the INR is therapeutic on warfarin. Note that rivaroxaban can contribute to INR elevation or,
- Overlap warfarin with rivaroxaban until the INR is therapeutic on warfarin, testing right before the next rivaroxaban dose to minimize the effect of rivaroxaban on INR elevation.
Start the second DOAC when the next dose of the first DOAC would have been due. Do not overlap.
Protamine sulfate 1 mg per 100 anti-Xa units if LMWH administered within last 9 hours. If ineffective, may give additional protamine sulfate 0.5 mg per 100 anti-Xa units.
The VKAs produce their anticoagulant effect by interfering with the cyclic interconversion of vitamin K and its 2,3 epoxide (vitamin K epoxide), thereby modulating the gamma-carboxylation of glutamate residues (Gla) on the N-terminal regions of vitamin K-dependent proteins, which include factors II, VII, IX, and X. The vitamin K-dependent coagulation factors require gamma-carboxylation for their procoagulant activity. Carboxylation is required for a calcium-dependent conformational change in coagulation proteins that promotes binding to cofactors on phospholipid surfaces. In addition, the VKAs inhibit carboxylation of the regulatory anticoagulant proteins C, S, and Z and thereby have the potential to be procoagulant.
Read more here.
1 unit vWF:RCo/kg infused will result in recovery of 1.5-2 units vWF:RCo/dL in adults; 50 units vWF:RCo/kg will give a recovery of 75-100 units vWF:RCo/dL.
DDAVP increases the release of vWF and factor VIII from endothelial cell stores by releasing vWF from cytoplasmic organelles called Weibel-Palade bodies.
From Levy and Connors:
Heparin polymers bind to antithrombin and thereby accelerate the interaction between antithrombin and thrombin or antithrombin and factor Xa, either of which results in the inhibition of prothrombotic activity. A specific pentasaccharide sequence of unfractionated heparin and low-molecular-weight heparin binds to antithrombin and can inhibit factor Xa, but longer, unfractionated heparin polymers containing 18 or more polysaccharide units are required to inhibit thrombin. A specific pentasaccharide sequence of unfractionated heparin and low-molecular-weight heparin binds to antithrombin and can inhibit factor Xa, but longer, unfractionated heparin polymers containing 18 or more polysaccharide units are required to inhibit thrombin.
Learn more here
LMWH is derived from full-length, highly sulfated polysaccharide (unfractionated heparin) harvested from porcine mucosal tissue by chemical or enzymatic depolymerization, resulting in smaller size polysaccharide fragments (6-22 sugar residues). Heparin binds to antithrombin through a pentasaccharide region leading to the inhibition of thrombin and Factor Xa (FXa). . Only one third of heparin polysaccharide chains display anticoagulant properties implying that a specific sequence in the polysaccharide chain is required for this activity. The number of polysaccharide fragments determines which serine proteases are inactivated by antithrombin. Inactivation of thrombin requires that the heparin be long enough (> 18 sugar residues) to bind both antithrombin and thrombin, serving as a bridge between the protease and its inhibitor. By contrast, inactivation of FXa simply requires the pentasaccharide sequence to bind to antithrombin (there is no need for bridging FXa with antithrombin). The majority of LMWH is shorter than this threshold. Thus, most of its activity is exerted through factor Xa inhibition.
Related FAQs:
- What is the half-life of low-molecular-weight heparin (LMWH)?
- Is low-molecular-weight heparin (LMWH) safe in patients with chronic liver disease/cirrhosis?
- How do I use protamine sulfate to reverse low-molecular-weight heparin (LMWH)?
- Does treatment of venous thromboembolism (VTE) with low-molecule-weight heparin (LMWH) require one or two doses per day?
- What is the renal clearance of LMWH?
- What does LMWH stand for?
- How is low-molecular-weight heparin (LMWH) monitored?
Vitamin K antagonists disrupt the carboxylation of vitamin K-dependent coagulation factors (factors II, VII, IX, and X; and protein C and protein S). Administration of vitamin K restores hepatic carboxylation of the vitamin K-dependent coagulation factors in a dose-dependent manner. Since this requires new protein synthesis, administration of vitamin K does not result in immediate correction of coagulopathy. 12-24 hours are required for a sufficient amount of newly synthesized carboxylated vitamin K-dependent coagulation factors to reconstitute hemostasis.
Learn more here.
Related FAQs:
Immediate onset of action, which lasts for about 12-24 hours.
Related FAQs:
Thrombosis occurs in one-third to one-half of patients with HIT. It may occur in veins, arteries or microvessels.
About 25% of all VTE events reported to be due to recurrent VTE.
aPTT; adjust to 1.5-3.0 times baseline.
aPTT; adjust to 1.5-2.5 times baseline.
A condition in which the blood’s ability to clot is impaired. .
DDAVP may be administered IV, subcutaneously, or by nasal spray.
No one laboratory test is specific for DIC. Clinical practice guidelines recommend using a clinical scoring system, such as the ISTH scoring system, which includes consideration of the platelet count, D-dimers, fibrinogen and the prothrombin time.
Subcutaneously, once daily. Learn more here.
The need for high heparin doses to achieve a targeted level of anticoagulation. Some define it as the need for > 35,000 U heparin per day, or in the setting of cardiopulmonary bypass, the need for a dose of more than 500 U per kilogram of
body weight to achieve an activated clotting time of 400 to 480 seconds.
Learn more here
≥ 35,000 units/day of unfractionated heparin needed to achieve therapeutic aPTT. Learn more here.
Based on clinical presentation (thrombocytopenia and/or thrombosis in temporal association with heparin therapy without other obvious causes) plus presence of platelet-activating antiplatelet factor 4 (PF4)/heparin antibodies. Clinical prediction scores, especially the 4T score, are helpful in estimating the pretest probability of HIT and determining whether heparin should be discontinued and if further testing for antibodies should be carried out.
LWMH is monitored using a chromogenic heparin anti-Xa assay, which measures the ability of heparin to inhibit the activity of a known amount of activated factor X (FXa) in the reagent. The reagent includes an excess of antithrombin so that heparin in the sample is rate-limiting reagent for Xa inhibition. Heparin in the patient sample inhibits the enzymatic conversion of a Xa-specific chromogenic substrate to colored product by factor Xa (residual Xa is assayed using a chromogenic substrate specific for Factor Xa). Standard curves are created using multiple concentrations of UFH and LMWH and are used to calculate concentration in the patient plasma. Samples are obtained 4 hours following subcutaneous injection of LMWH. Therapeutic levels for enoxaparin (Lovenox) are typically 0.60-1.00 IU/mL for twice daily dosing.
- Dose calculated based on the dose of heparin administered and the elapsed time since the last heparin dose.
- 1 mg protamine sulfate IV neutralizes about 100 units of unfractionated heparin (UFH) given in past 2-3 hours (for example, protamine sulfate 25-35 mg would be administered in a patient receiving 1,000-1,250 units/hour UFH).
- Given slowly over 10 minutes at a rate not to exceed 5 mg/minute.
- Dose should not exceed 50 mg. When given at high dose, protamine sulfate can cause severe hypotension, cardiovascular collapse, noncardiogenic pulmonary edema, catastrophic pulmonary vasoconstriction, and pulmonary hypertension
Related FAQs:
- One-third of the drug is eliminated in an unchanged form through the kidney.
- Two-thirds metabolized by the liver and eliminated (about equally) through the kidney and gut.
- Mild, factor VIII activity > 5%-40%
- Moderate, factor VIII activity 1%-5%
- Severe, factor VIII activity < 1%


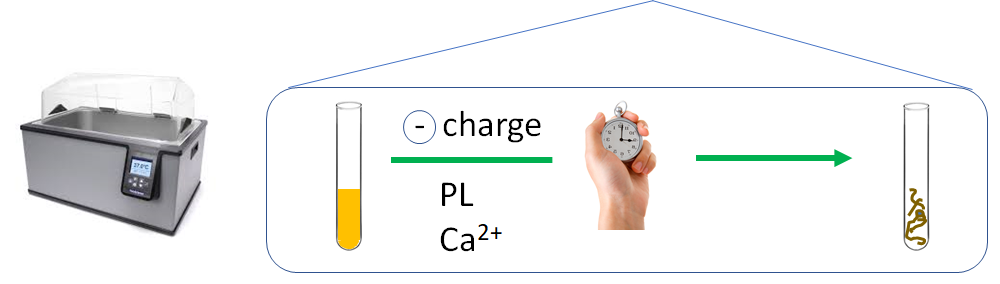
Calculated from the PT ratio (patient PT/control PT) adjusted for the international sensitivity index (ISI); ISI derived from a cohort of patients on stable anticoagulation with vitamin K antagonists.
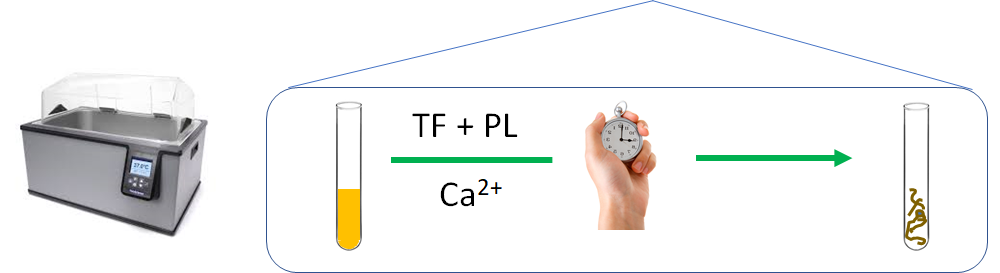
Two options: 1) weight based – 80 units/kg bolus followed by 18 units/kg/hour infusion, 2) fixed dose – bolus of 5,000 units followed by 1,000 units/hour.
ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease:

Protamine sulfate
About 6 days because factor II (prothrombin) has a long half-life.
The vitamin K family is comprised of multiple similarly structured fat-soluble molecules containing a 2-methyl-1,4-naphthoquinone ring structure called menadione. There are three main structures of vitamin K:
- Natural occurring:
- Phylloquinone (K1)
- The predominant form of vitamin K present in the diet.
- Predominantly found in green vegetables and plant chlorophylls.
- Used to treat patients with vitamin K deficiency or to reverse vitamin K antagonists.
- Menaquinones (K2)
- Further subdivided into:
- Short-chain (e.g., menaquinone-4; MK-4)
- Long-chain (e.g.,, MK-7, MK-8, and MK-9)
- Synthesized by bacteria.
- Primarily found in food where bacteria are part of the production process.
- Further subdivided into:
- Phylloquinone (K1)
- Synthetic origin:
- Menadione (K3)
- Converted into K2 in liver.
- Harmful effects shown in humans, therefore not used therapeutically.
Both vitamin K1 and K2 can function as cofactors in the carboxylation process of vitamin K-dependent proteins.
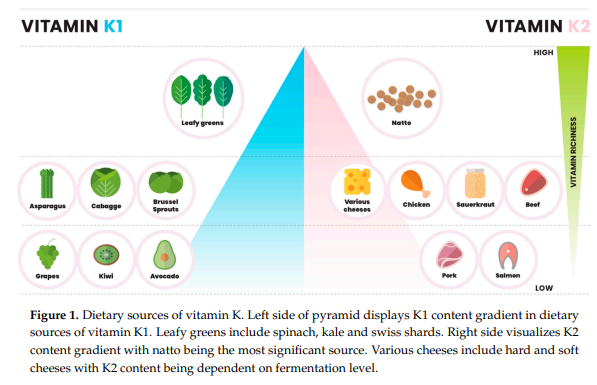
Learn more here.
Related FAQs:
About 12 million in the United States!
About 30% of patients within 10 years.
About 30% of patients
The current classification includes types 1 and 3, which are characterized by quantitative deficiencies of von Willebrand
factor (VWF), as well as types 2A, 2B, 2M, and 2N, which are qualitative variants.
Three main types are:
- Type 1 – partial quantitative deficiency of vWF.
- Type 2 – qualitative abnormalities of VWF; 4 subtypes:
- 2A – characterized by reduced or absent high-molecular-weight vWF.
- 2B – gain of function in VWF that increases its affinity for platelets.
- 2M – caused by reduced VWF interactions with platelets or collagen.
- 2N – results from reduced binding of VWF to FVIII.
- Type 3 – virtual absence of the VWF protein with associated very low FVIII levels.
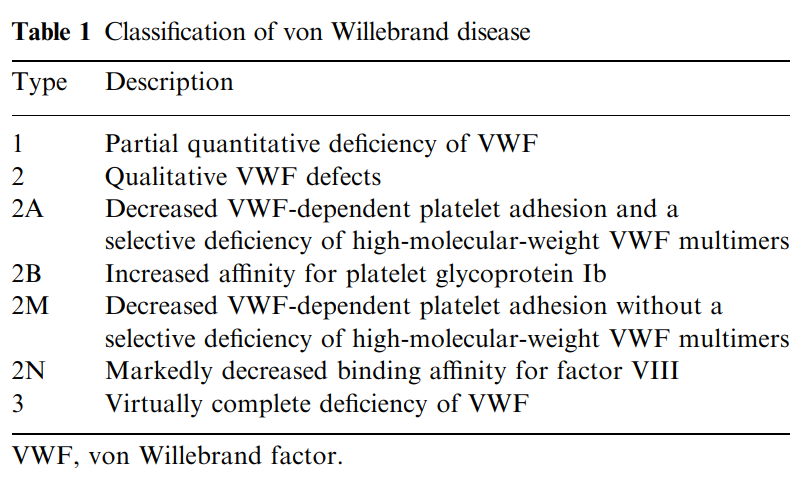
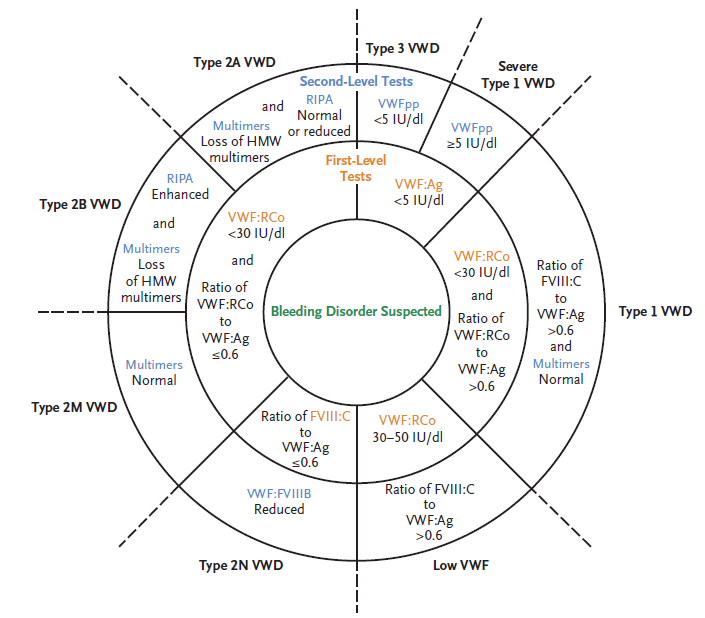
25%-40% of cases
10-20% of patients
Start initial treatment with parenteral anticoagulation. Administer VKA early (for example, same day as start of parenteral therapy) and continue parenteral anticoagulation for ≥ 5 days and until INR is ≥ 2 for at least 24 hours. Learn more here.
Vitamin K antagonist is preferred over low-molecular weight heparin (LMWH).

Once the platelet count is over 150 x 109/L, switch to warfarin (with appropriate overlap) or preferably to a DOAC (see 2018 American Society of Hematology [ASH] guideline recommendation 3.9 below). In case of HIT without thrombosis, continue for no longer than 3 months (typically 4-6 weeks). In the case of HIT complicated by thrombosis (termed HITT), treat for 3-6 months (see ASH guideline recommendation 3.8 below).


Even with platelet count recovery, patients remain at risk for thrombosis for 4 to 6 weeks after diagnosis (~17–53% over a thirty day period) because of circulating anti-PF4/heparin antibodies. While there is consensus among experts that patients with isolated HIT should be treated in the short-term with alternative anticoagulants, there is no firm consensus on the intensity or duration of anticoagulation for this patient population.
Platelet count recovery occurs within 7 days in 90% of cases. Functional assays become negative at a median of 50 days after heparin is suspended, while circulating anti-PF4/heparin antibodies are no longer detectable by immunoassay at a median of 85 days.
Median 50 days for platelet activation assays and 85 to 90 days for immunoassays.
No. For patients who have received heparin in the last 100 days, thrombocytopenia can develop rapidly (median = 10.5 hours), due to circulating PF4/H antibodies. However, patients who are re-exposed to the drug months to years after antibody disappearance do not manifest anamnestic responses, and seroconversion risk appears similar to de novo heparin exposure.
If the clot extends proximally, especially into the proximal veins. The recommendation to anticoagulate if the clot is found to extend within the distal veins is weaker.
2021 ACCP clinical guideline recommendations:

Day of first heparin exposure
Highest platelet count that immediately precedes the putative HIT-related fall in platelet count.
- < 72 hours post-surgery
- Infection (confirmed bacteremia/fungemia)
- Chemotherapy or radiation < 20 days
- Disseminated intravascular coagulation (DIC)
- Posttransfusion purpura
- Other drugs implicated in drug-induced thrombocytopenia
No. There are 4 parameters that are used in clinical scoring systems for DIC: fibrinogen, prothrombin time (PT), platelet count and D-dimers/fibrin degradation products. Fibrinogen is the least sensitive of these markers (overall sensitivity of low fibrinogen level reported to be 28%) because it is an acute phase reactant and therefore often elevated in conditions associated with DIC.
No, unless a decision has been made to stop anticoagulation in such patients.
2019 ASH guideline recommendations:

2021 ACCP clinical guideline recommendations:


No, it is a complication of an underlying diseases such as sepsis or cancer.
No, all are associated with similar reduction in VTE recurrence.
The aPTT may be increased in Type 2N vWD (the mutation specifically impairs the ability of vWF to carry FVIII) and Type 3 vWD (since vWF carries and stabilizes FVIII in the circulation, the very low levels of vWF associated with Type 3 vWD result in low FVIII activity).
No. Below are the 2019 ASH guideline recommendations:
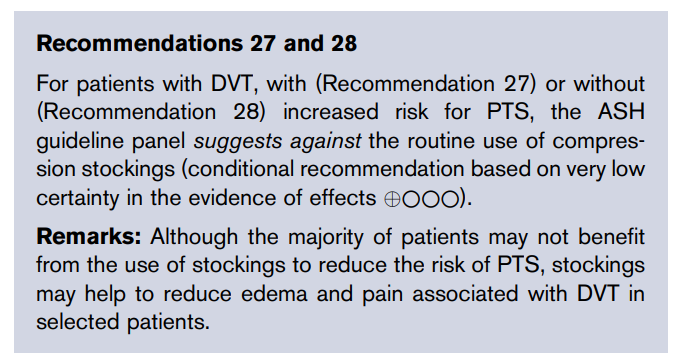
Yes, It’s called Vonvendi.
- Consider idarucizumab.
- Consider oral activated charcoal to prevent further absorption of dabigatran if last dose was taken in the last 2 hours.
- If idarucizumab is not available, consider:
- 4-factor PCC or activated PCC (aPCC).
- Hemodialysis, hemofiltration, or charcoal hemoperfusion to facilitate further clearance of dabigatran, particularly in patients with renal failure.
- Consider andexanet alfa.
- If andexanet alfa is not available, consider 4-factor PCC 50 units/kg IV.
- If 4-factor PCC is not available, consider activated PCC 50 units/kg IV.
- Consider activated charcoal for known ingestion within last 2-4 hours.
- Stop anticoagulant.
- Assess for and manage comorbidities that might contribute to bleeding, such as, thrombocytopenia, uremia, or liver disease.
- Provide supportive care with volume resuscitation (IV fluids) and hemodynamic support (inotropes, monitoring) as needed.
- Initiate local or surgical hemostatic measures to control bleeding.
- Red blood cell transfusion as needed.
- Administer vitamin K 5-10 mg IV
- Consider administering 4-factor prothrombin complex concentrate (PCC):
- 25 units/kg for INR 2-4
- 35 units/kg for INR 4-6
- 50 units/kg for INR > 6
- Recombinant factor VIIa (rFVIIa) is not recommended.
Guideline recommendations:
American Society of Hematology 2018 guidelines:


2012 American College of Chest Physicians Evidence-Based Clinical Practice Guidelines:


If INR is > 10 and the patient is not bleeding, consider withholding 2 doses and administering oral vitamin K 2.5-5 mg. Subsequent dosing should be decreased by 15%-20%.
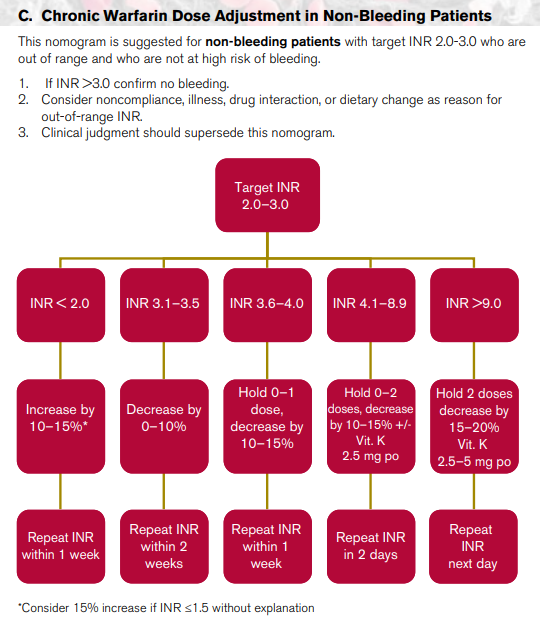
No, routine use of vitamin K is not suggested in patients with INR 4.5-10.
Tranexamic acid is 6- to 10-fold more potent compared with aminocaproic acid.
Dabigatran and edoxaban (parenteral anticoagulation administered for 5-10 days before).
No, but ABO compatibility is preferred.
Graduated compression stockings do not reduce the incidence of post-thrombotic syndrome but may provide symptomatic relief from DVT symptoms. Routine use of compression stockings to prevent PTS in patients with acute DVT of the leg is not recommended, though they may be considered in patients with symptomatic swelling.
American Society of Hematology 2020 guidelines for management of venous thromboembolism: treatment of deep vein thrombosis and pulmonary embolism:

Yes, bilateral dopplers of the lower extremities are recommended in patients with acute isolated HIT.

Only if the patient has an upper-extremity central venous catheter, or has signs or symptoms suggestive of upper extremity DVT.

Not clear whether anticoagulation therapy is beneficial in this low risk case, but clinical practice guidelines recommend treating with anticoagulant if patients are highly symptomatic or if there is a high risk for extension. The alternative is to withhold anticoagulation and carry out serial imaging for 2 weeks to rule out extension.
Routine administration of vitamin K is not recommended to increase the plasma levels of coagulation factors in patients with cirrhosis.
From the Italian Association for the Study of Liver Diseases (AISF) and the Italian Society of Internal Medicine (SIMI) 2016 guideline:

No, it is not routinely recommended.
Patients can be treated at home provided they are feeling well enough to be at home and have the necessary support.
2019 ASH guideline recommendations:

Not necessarily. In cases where the risk for clot extension is not high, they may be followed by serial doppler imaging of the legs.
For example, 2021 ACCP clinical guideline recommendations:


No, not unless there is another indication for these drugs, for example a coronary stent, and provided the risk: benefits are acceptable.

No, platelet transfusions may exacerbate the thrombotic tendency in HIT. Platelet transfusion should be reserved for patients with active bleeding or at high risk of bleeding.

No. Patients should be encouraged to ambulate early, if possible.
Patients without antiphospholipid syndrome should receive a DOAC. DOACs are preferred over vitamin K antagonists such as warfarin/coumadin.
From the 2019 ASH guideline:


2021 ACCP clinical guideline recommendations:



- Immunoassays that detect presence of antibodies to platelet factor (anti-PF4/heparin antibodies).
- Functional assays – such as the serotonin release assay – that detect anti-PF4/heparin antibodies capable of activating platelets.
- Argatroban
- Bivalirudin
- Danaparoid
- Fondaparinux – especially in clinically stable patients at average risk of bleeding, and provided normal renal function.
- DOAC:
- Especially in clinically stable patients at average risk of bleeding.
- Most of the published experience in HIT is with rivaroxaban.

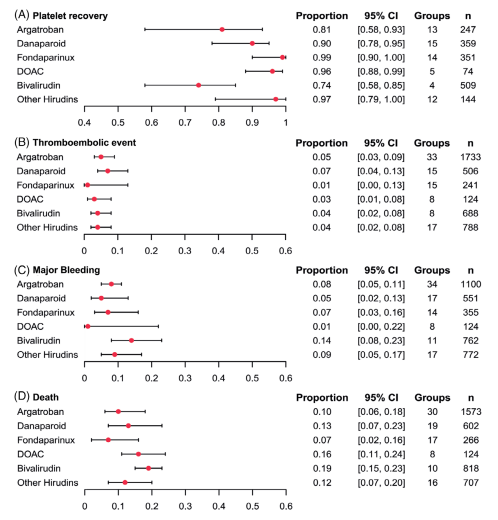
Autoantibodies that deplete or inhibit activity of target coagulation factors (most commonly factor VIII) leading to development of bleeding complications. Learn more here.
- Anaphylaxis in patients previously been exposed to protamine, including diabetic patients who have received protamine-containing insulin (eg, NPH, PZI), and individuals with fish allergy (protamine is derived from fish).
- Boxed Warning includes the following adverse events in patients who have previously received protamine sulfate or other protamine-containing drugs (e.g., NPH insulin, protamine zinc insulin, and certain beta-blockers):
- Hypotension
- Cardiovascular collapse
- Non-cardiogenic pulmonary edema
- Catastrophic pulmonary vasoconstriction
- Pulmonary hypertension
- Thrombosis events – reported in 5%-10% (arterial and venous)
- Anaphylaxis and hypersensitivity reactions

Deficiency or inhibitor of one or more components of the intrinsic pathway including contact pathway factor (especially FXII, but also HMWK and prekallikrein), FXI, FIX, and FVIII, or antibodies against phospholipids (lupus anticoagulant).
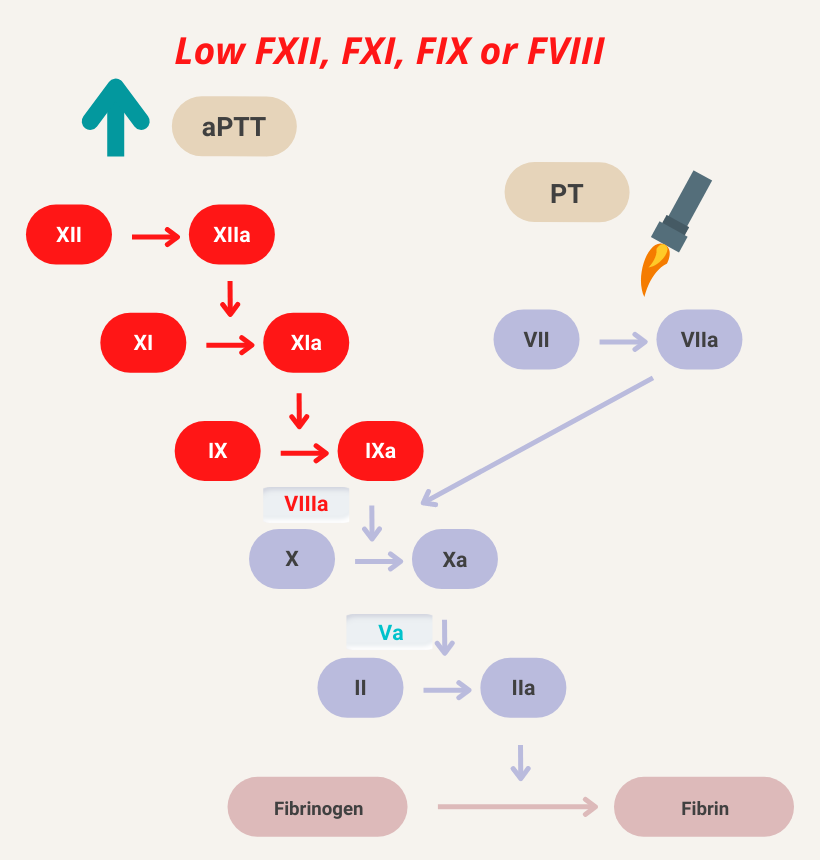
Factor VII (FVII) deficiency, FVII inhibitor (rare), early vitamin K deficiency, vitamin K antagonist, chronic liver disease, and disseminated intravascular coagulation (DIC).
Liver disease and DIC affect multiple clotting factors in the extrinsic, intrinsic and common pathway. These conditions may be associated with isolated elevation of the PT because FVIII levels are often high (acute phase reactant, produced in endothelial cells), compensating for depletion of other factors in the intrinsic pathway.
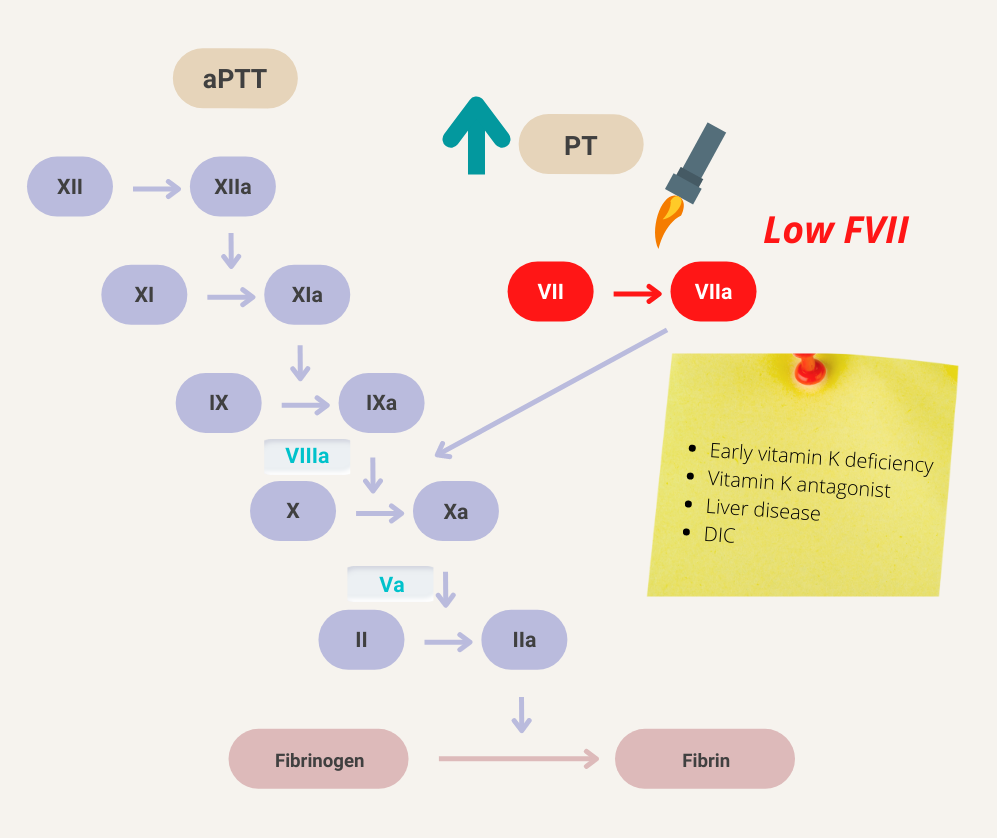
Inflammation (FVIII is an acute phase reactant), pregnancy, increased levels of von Willebrand factor (vWF; which binds and stabilizes FVIII) and, rarely, reduced clearance.
Deficiency or inhibitor of common pathway factors (factor X, factor V and prothrombin) or combined deficiencies of intrinsic and extrinsic pathways (for example chronic liver disease, heparin treatment, and disseminated intravascular coagulation [DIC]).
- Non-specific binding of negatively charged heparin molecules to:
- Platelet factor 4
- Histidine-rich glycoprotein
- Lipoproteins
- von Willebrand factor
- Factor VIII
- Fibrinogen
- Monocytes
- Endothelial cells
- Growth factors
- Nonendothelial surfaces
- Intravenous tubing
- Extracorporeal circuit components
- Deficiency of antithrombin
- Congenital
- Acquired
- Liver disease
- Sepsis
- Acute disseminated intravascular coagulation
- Asparaginase use in patients with acute leukemia
- Use of extracorporeal circuits in cardiopulmonary bypass or ECMO
Learn more here
Aminocaproic acid and tranexamic acid
- Degradation product of cross-linked fibrin.
- The smallest circulating fibrin degradation product that is specific for fibrinolysis (as distinct from fibrinogenolysis).
- Contains two cross-linked D fragments of the fibrin protein, hence its name.
- High levels in patients with disseminated intravascular coagulation (DIC), but high levels can also be found in patients with venous thromboembolism, recent surgery, or inflammatory conditions.
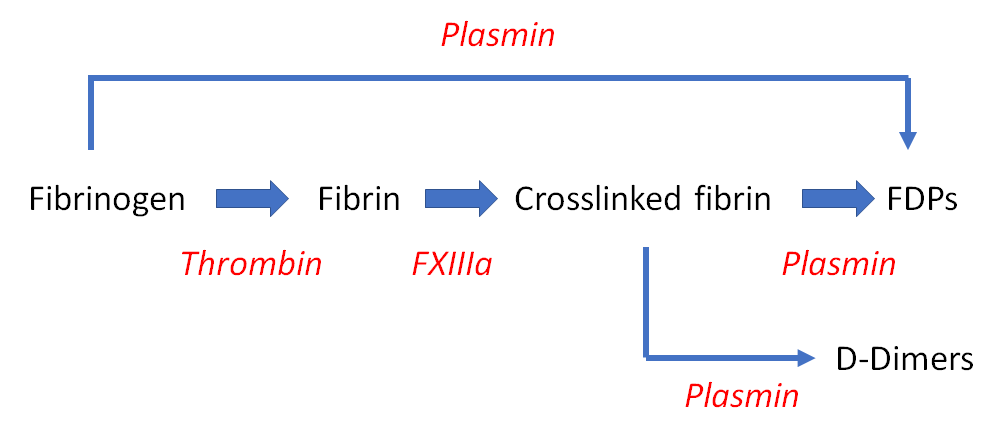
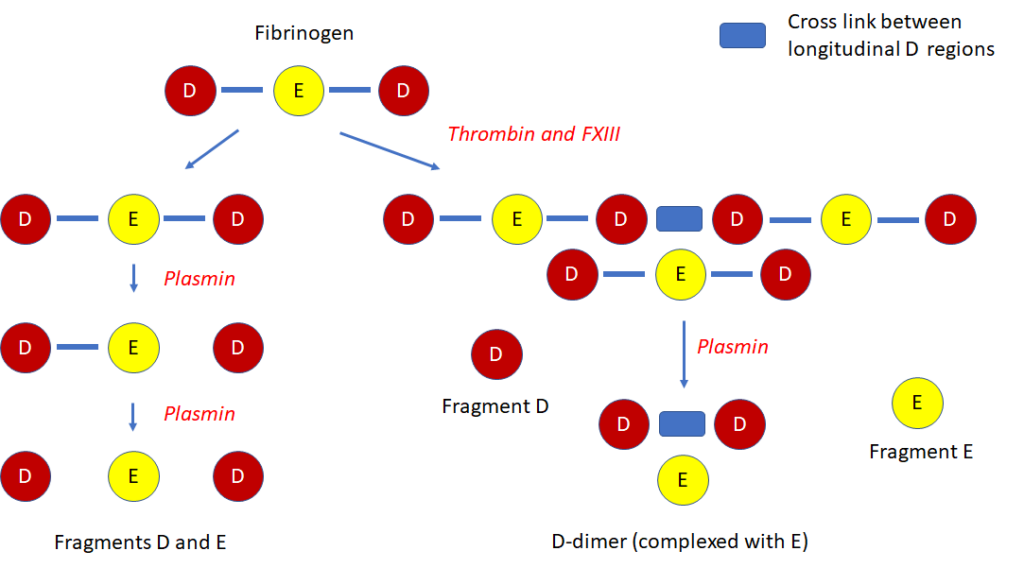
- Warfarin – half-life 36 h
- Acenocoumarol – half-life 10 h
- Phenprocoumon – half-life 5 days
- Fluindione – half-life 56 days; used mostly in France
- Anisindione – half-life not known
- Treatment of bleeding episodes and peri-operative management in adults and children with hemophilia A or B with inhibitors, congenital factor VII (FVII) deficiency, and Glanzmann’s thrombasthenia with refractoriness to platelet transfusions, with or without antibodies to platelets.
- Treatment of bleeding episodes and peri-operative management in adults with acquired hemophilia.
- von Willebrand factor antigen (vWF:Ag)
- von Willebrand factor ristocetin cofactor activity (vWF:RCo) (platelet-binding activity of vWF measured by vWF:RCo assay)
- Factor VIII activity (FVIII:C)
- von Willebrand factor collagen binding (vWF:CB) activity (recommended by BCSH but not NHLBI as first-level test)
In patients with normal renal function:
- Rivaroxaban – half-life 5-9 hours
- Apixaban – half-life 11.7-13.5 hours
- Dabigatran – half-life 14-17 hours
If cirrhosis is decompensated, consider anticoagulation in patients who are candidates for liver transplantation, those with symptoms, and those in whom the thrombus extends into the superior mesenteric vein leading to suspected intestinal ischemia.
If cirrhosis is compensated, anticoagulation is recommended (though the decision should be individualized). Anticoagulation is for at least 6 months. Before starting anticoagulation, consider screening for varices. Acute development of portal vein thrombosis favors anticoagulation therapy.
Guideline recommendations
2020 ACG Clinical Guideline: Disorders of the Hepatic and Mesenteric Circulation:

2020 Practice Guidance by the American Association for the Study of Liver Diseases:





Conditions associated with fibrinogen consumption and/or loss or fibrinogen dysfunction. Examples include major bleeding, postpartum hemorrhage, disseminated intravascular coagulation (DIC), and cardiac surgery.
- von Willebrand factor antigen (vWF:Ag)
- von Willebrand factor ristocetin cofactor (vWF:RCo) activity
- FVIII activity
Learn more here.
- Unfractionated heparin > low molecular weight heparin (these differences are especially true at prophylactic doses)
- IV heparin > SC heparin
- Therapeutic or prophylactic dose heparin > heparin flush
- Bovine>porcine heparin
- von Willebrand factor (vWF) multimer assay
- Ristocetin-induced platelet aggregation (RIPA)
- vWF propeptide (vWFpp)
- vWF factor VIII binding (vWF:FVIIIB)
Systemic infection, solid and hematological malignancies, obstetric diseases (for example, abruptio placentae or amniotic embolism), trauma, aneurysms, and liver diseases.

Headache, facial flushing, tachycardia, and hyponatremia
Thrombotic type DIC and fibrinolytic type DIC.
Soleal muscle veins connected with posterior tibial or peroneal veins and gastrocnemius muscle veins that drain into the popliteal vein. Thrombosis of these veins is termed isolated calf muscle vein thrombosis [ICMVT], and is treated the same as other forms of distal DVT.
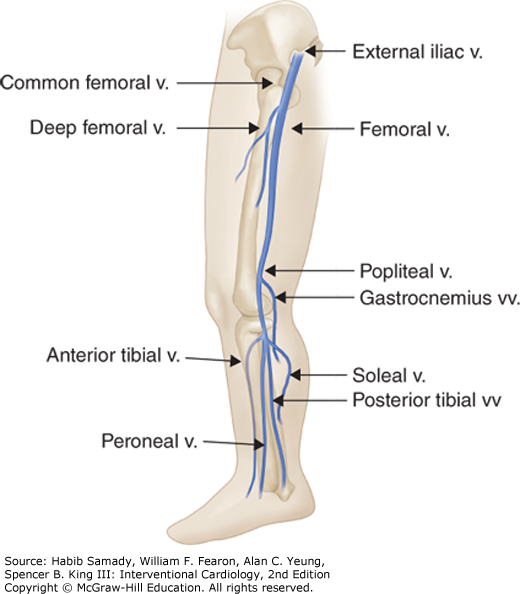
Thromboelastography (TEG) and rotational thromboelastometry (ROTEM). The following is a helpful description of the similarities and differences between the two. “Whole blood (a minute amount of it, no more than 1 ml) at body temperature (37º) is added to a heated cuvette (a little cup). A pin is suspended into the cup, and then some sort of rotation takes place. In fact the main difference between TEG and ROTEM is the bit which rotates (TEG rotates the cup, and ROTEM rotates the pin). Irrespective of which bit is rotating, some impediment to the rotation develops as the blood clots. The degree of this impediment is recorded as “amplitude”, and displayed on the time vs. amplitude graph.” An example of the type of graph generated by these instruments is shown here (the details are not important in the context of this short answer).
Faster reconstitution, ease of use, and not requiring thawing or ABO compatibility.
Causes of heparin resistance include:
- Antithrombin (AT) deficiency (≤60% levels)
- Increased heparin clearance
- Increased levels of heparin-binding proteins
- Elevated levels of FVIII
- Elevated levels of fibrinogen
- Medications such as aprotinin
- Thrombocytosis
Learn more here.
Unexplained extremity swelling, pain, warmth, or erythema.
- Non-activated PCCs (PCCs):
- Nonactivated 3-factor PCCs which contain factors II, IX, and X, with negligible factor VII, protein C, and S.
- Nonactivated 4-factor PCCs which contain factors II, VII, IX, X, and proteins C and S.
- Activated PCCs (aPCCs):
- Also called factor VIII inhibitor bypassing agent.
- Contains small amounts of factor IXa, Xa, and thrombin and large quantities of factor VIIa.
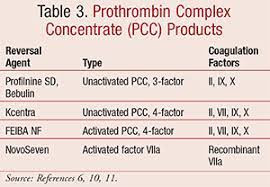
Prevent pulmonary embolism, recurrence of DVT and occurrence of post-thrombotic syndrome.
ISTH/SSC Harmonization of the recommendations:
- Fresh frozen plasma (FFP) recommended in patients with active bleeding or requiring an invasive procedure and either prolonged PT/aPTT (> 1.5 × normal) or decreased fibrinogen (< 1.5 g/L).
- Cryoprecipitate recommended for patients with active bleeding plus severe hypofibrinogenemia (< 1.5 g/L) that persists despite FFP replacement.
- Platelet transfusion recommended in patients with active bleeding plus platelet count < 50 × 109/L, or high risk of bleeding plus platelets < 20 × 109/L.
- Unfractionated heparin (UFH)
- Low-molecular-weight heparin (LMWH)
- Fondaparinux
- Argatroban
- Bivalirudin
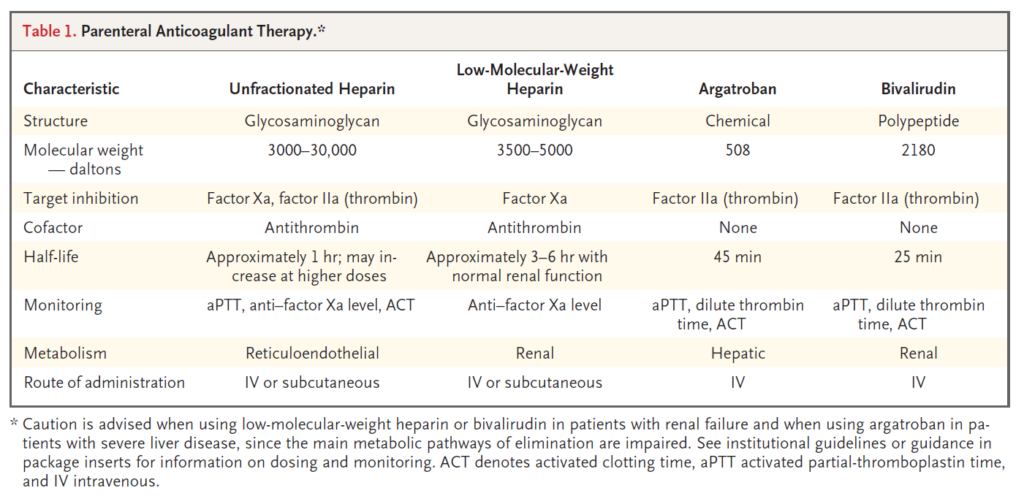
Atypical HUS and infection-induced HUS, including Shiga toxin-producing Escherichia coli-associated (STEC) HUS and S. pneumoniae-associated HUS.
Peroneal veins (paired), posterior tibial vein and anterior tibial vein.
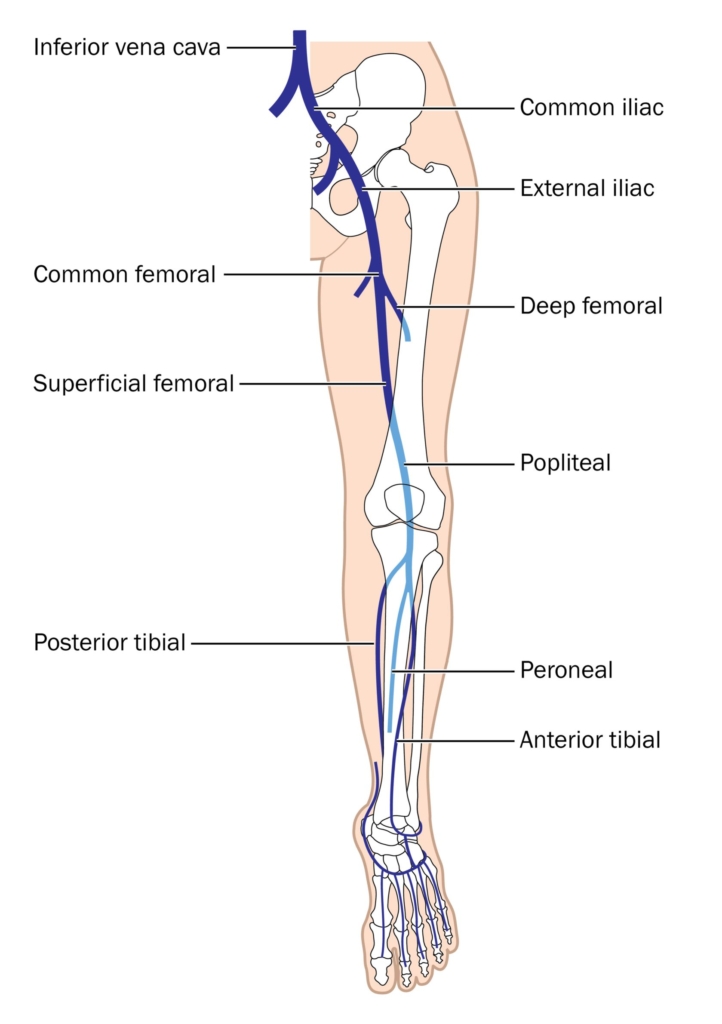
3 pillars of treatment are:
- Treat underlying disease that triggered DIC.
- Provide replacement therapy where appropriate.
- Inhibit thrombin and fibrin formation where appropriate.
- Type 1 – 75%
- Type 2 – 20%
- Type 3 – 5%
- Tachycardia
- Tachypnea
- Pleural rub
- Loud P2 (pulmonic valve closure sound)
- Neck vein distention
- Arterial hypotension and shock (in severe cases)
- Type 2A – decreased secretion or increased cleavage of high-molecular-weight multimers.
- Type 2B – increased vWF binding to platelet glycoprotein Ib (GPIb) alpha receptor with rapid clearance of platelet-vWF complex.
- Type 2M – defective binding to platelet GPIb alpha receptor or collagen; M for multimer.
- Type 2N – defective vWF binding to factor VIII; N for Normandy, where it was discovered.
Learn more here.
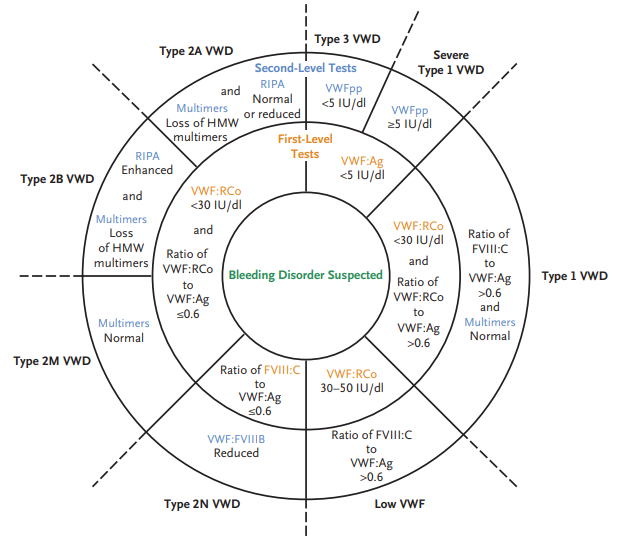
- Initiation phase: administration of anticoagulant after diagnosis of DVT, and consisting of parenteral or full dose oral anticoagulation for 5-21 days, depending on anticoagulant used.
- Treatment phase: administration of anticoagulation following initiation phase, consisting of anticoagulation administered at standard therapeutic doses and considered complete after 3 months of anticoagulation.
- Extended phase: administration of anticoagulants without pre-planned stop date, at full or reduced dose, as secondary prevention (to reduce recurrent DVT).
Another perspective:
Vitamin-dependent procoagulants include factors II, VII, IX, X. Vitamin-dependent anticoagulants include protein C and protein S.
A group of disorders characterized by microangiopathic hemolytic anemia, thrombocytopenia and microthrombi leading to ischemic tissue injury. Laboratory tests reveal thrombocytopenia and hemolytic anemia with red blood cell fragmentation (schistocytes) on the peripheral blood smear.
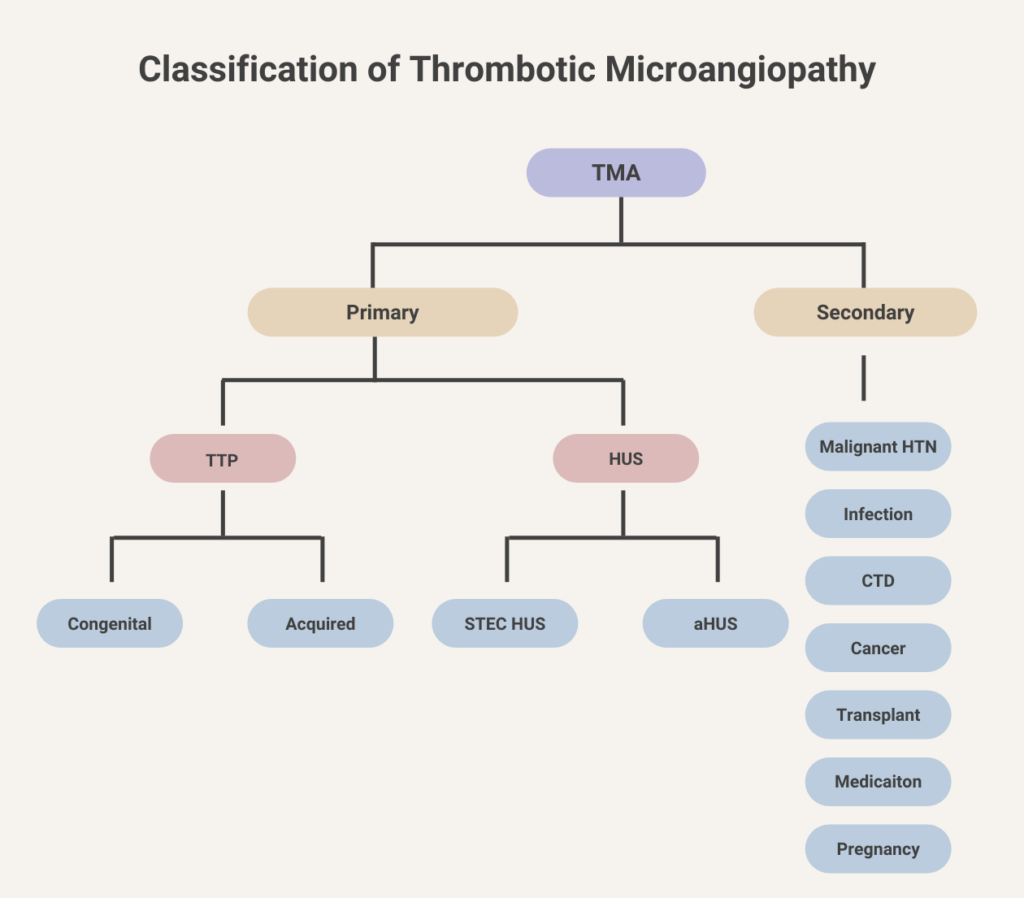
Point-of-care assays used to assess all phases of coagulation, providing quantitative graphic measure of platelet function, clotting strength, and fibrinolysis.
About 25% lower than in persons with other blood types.
Aging, pregnancy, inflammation (vWF is an acute phase reactant)
Fibrinogen, von Willebrand factor (vWF), and factors VIII and XIII.
Factors II, VII, IX, X
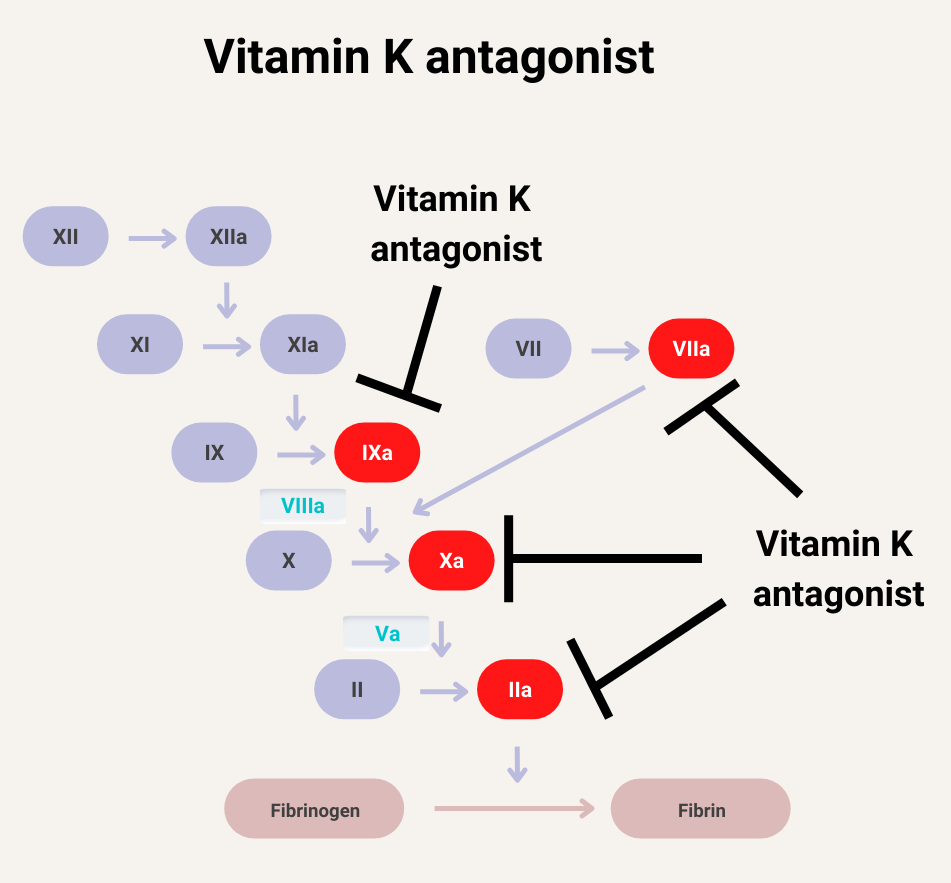
All except factor VIII, which is synthesized by endothelial cells in the liver and elsewhere in the body.
- Aging
- Pregnancy
- Stress (acute phase reactant)
- African Americans (vWF:Ag levels 20% higher compared to white patients, but vWF:RCo not affected)
Discontinue non-heparin anticoagulant and resume heparin.
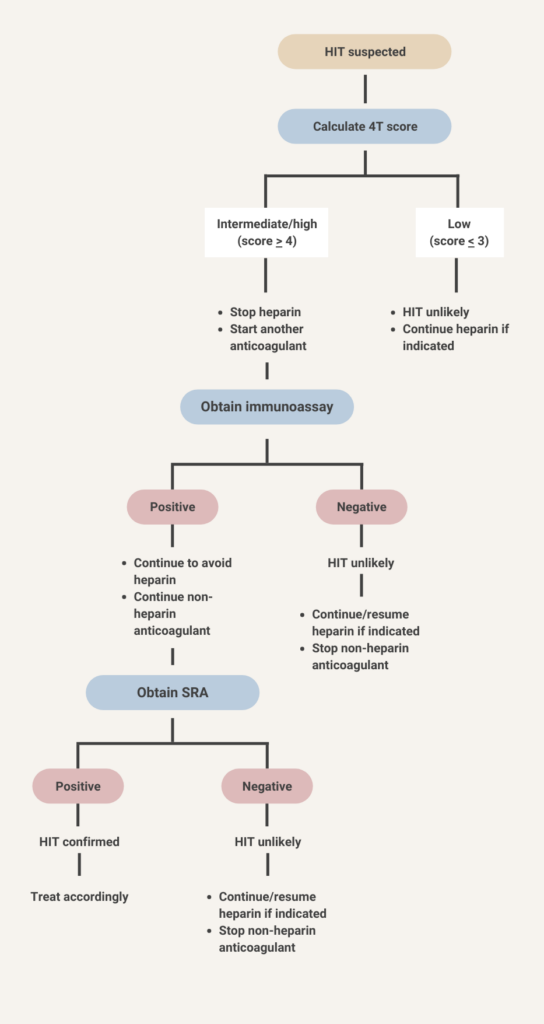
- Continue heparin.
- No need for alternative anticoagulant.
- HIT antibody testing not recommended.
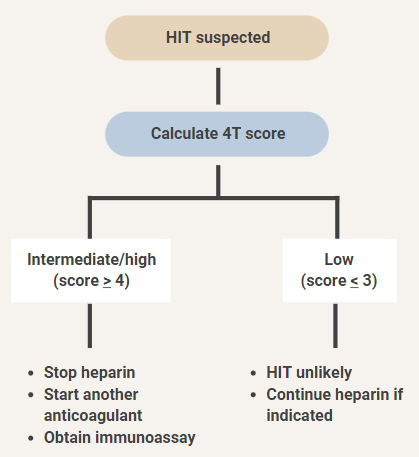
- Discontinue heparin.
- Initiate a non-heparin anticoagulant at therapeutic intensity.
- Test for HIT antibodies.
Anti-PF4/heparin antibodies of all isotypes (IgG, IgA, IgM, or polyclonal or polyspecific assays) or IgG isotypes.
1-desamino-8-D-arginine vasopressin, also called desmopressin
Disseminated intravascular coagulation
Direct oral anticoagulant
Normal levels of coagulation factors, antithrombin, and ADAMTS13.
Heparin-induced thrombocytopenia
Heparin-induced thrombocytopenia with thrombosis
International normalized ratio
Low-molecular-weight heparin
Prothrombin complex concentrate
Integrity of the extrinsic pathway of the clotting cascade.
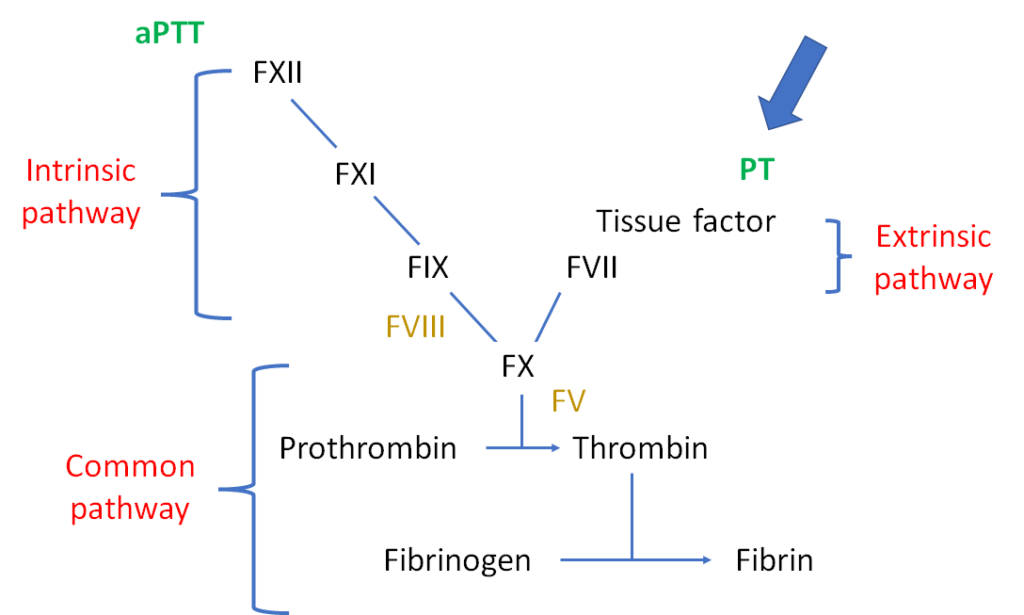
Postthrombotic syndrome
Rotational thromboelastometry. See image of ROTEM device from Werfen.
Thromboelastography, a type of viscoelastic test of whole blood clotting function. TEG contrasts with ROTEM, another type of viscoelastic test.
Thrombotic microagniopathy
Unfractionated heparin
Vitamin K antagonist
Venous thromboembolism
von Willebrand disease
Von Willebrand factor
Refers to DVT isolated to the deep veins below the knee. Compared with proximal DVT, distal DVT is associated lower rates of pulmonary embolism. Distal veins include paired peroneal veins, posterior tibial vein, and anterior tibial vein.
A transfusion (e.g. of fresh frozen plasma or platelets) to prevent bleeding.
A DVT caused by a clear trigger, for example, surgery, or bed confinement.
A DVT which extends into the popliteal vein or more proximally.
When there are contraindications to anticoagulation, particularly bleeding. Restarting or initiating anticoagulation is recommended if/when the bleeding risk resolves. Below are the 2019 ASH guideline recommendations:

2021 ACCP clinical guideline recommendations:

A transfusion (e.g. of fresh frozen plasma or platelets) to treat bleeding.
Alphanate is a high-purity factor VIII/von Willebrand factor (vWF) lyophilized concentrate used in patients with von Willebrand disease (vWD). it is FDA approved for adult and pediatric patients with vWD having surgery or invasive procedures in whom desmopressin (DDAVP) is ineffective or contraindicated.
A DVT that is not provoked by transient or persistent risk factors.
Modified recombinant factor Xa decoy protein that lacks procoagulant activity and binds to direct oral Xa inhibitors, including rivaroxaban and apixaban, low-molecular-weight heparins (LMWHs), and fondaparinux. FDA-approved in 2018 for reversal of rivaroxaban and apixaban when anticoagulation reversal is necessary due to life-threatening or uncontrolled bleeding.
Other information:
- Immediate onset with duration of activity of about 6 hours.
- Dosing regimens include:
- Low-dose regimen – initial IV bolus 400 mg at target rate of 30 mg/minute, followed by infusion at 4 mg/minute up to 120 minutes
- High-dose regimen – initial IV bolus 800 mg at target rate of 30 mg/minute, followed by infusion at 8 mg/minute up to 120 minutes
- Most common adverse effects include:
- Urinary tract infections
- Pneumonia
- Infusion-related reactions
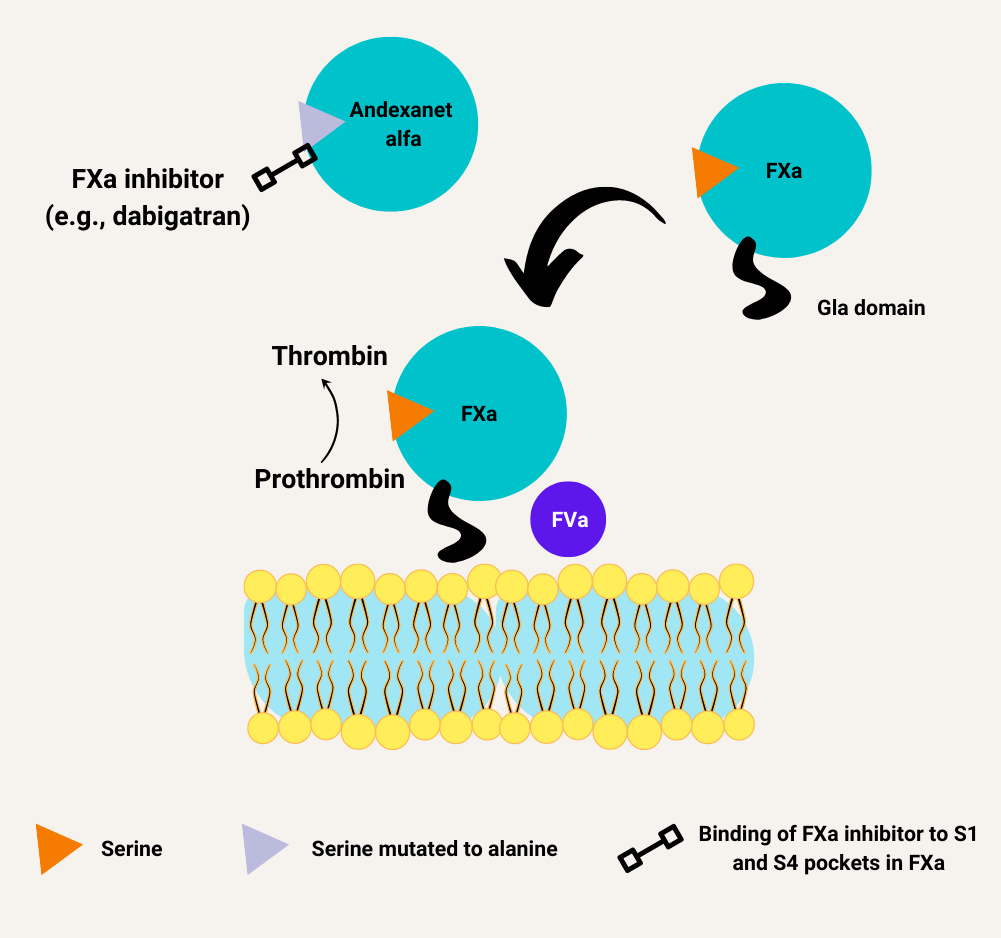
Learn more here.
Christmas disease
A type of thrombotic microangiopathy characterized by uncontrolled complement activation, microangiopathic hemolytic anemia, thrombocytopenia, and organ dysfunction, especially renal failure. Learn more here.
The administration of a short-acting anticoagulant before and after a procedure in a patient on anticoagulation, and then resumption of the anticoagulation therapy as soon as it is safe to do so following the procedure. Learn more here.
Hemophilia B (factor IX deficiency)
A byproduct of cryoprecipitate production generated after the removal of cryoprecipitate from thawed fresh frozen plasma centrifuged at 1-6 degrees C.
A blood product made by thawing fresh frozen plasma at 1-6 degrees C and recovering the cold insoluble precipitate by centrifugation. The precipitate is resuspended in a small quantity of plasma and refrozen/stored at at -18 to -25 degrees C.
HIT that develops or worsens after heparin has been discontinued. Thrombotic manifestations are delayed for days to weeks after heparin discontinuation and discharge.
According to Warkentin, TE:
There is a disorder called “delayed-onset HIT”. Despite its name, the timing of delayed-onset HIT resembles that of “typical onset HIT”, i.e. the platelet count fall begins 5–10 days after the immunizing exposure to heparin. Thus, there really is not any “delay” in onset of HIT (the term “delayed-onset” was intended to help the clinician remember that HIT can began several days after all heparin has been stopped). In recent years, the concept of delayed-onset HIT has expanded to include patients whose HIT worsens even after stopping heparin… patients’ HIT antibodies activate platelets in the absence of heparin, with activation inhibited by high heparin (100 IU/ml) and by Fcγ receptor blocking antibodies. These patients have a higher frequency of HIT-associated DIC, lower platelet count nadirs, and greater likelihood of severe sequelae, including venous limb gangrene. Sometimes, heparin “flushes” are the only proximate heparin exposure identified.
DDAVP is a synthetic derivative of antidiuretic hormone vasopressin that acts through type 2 vasopressin receptor.
Acquired clinicopathological syndrome characterized by systemic activation of coagulation that may cause organ-damaging thrombosis and/or hemorrhage due to consumption of coagulation factors and platelets. Defined by ISTH as “an acquired syndrome characterized by the intravascular activation of coagulation with a loss of localization arising from different causes; can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction”.
A vasomotor disturbance characterized by periodic attacks with triad of increased temperature, erythema, and burning pain resulting from hyperperfusion of skin areas, especially in the feet and hand. Typically relieved by cooling and aggravated by warming. May be primary or secondary. Causes of secondary erythromelalgia include polycythemia vera and essential thrombocythemia.
Learn more here.
Concentrated fibrinogen derived from pooled human plasma which is available as pasteurized and lyophilized powder.
Fondaparinux is a synthetic analog of pentasaccharide mimicking the site of heparin that binds to antithrombin and enhances the inactivation of factor Xa without interaction with factor II (prothrombin):
- Achieves steady state antithrombotic activity 3-4 days after administration.
- Almost completely dependent on kidneys for clearance; contraindicated in patients with severe renal impairment (creatinine clearance 30 mL/minute).
- Half-life ranges from 17 hours in young patients to 21 hours in older adults, and is further increased in patients with renal insufficiency.
- Predictable anticoagulant effect so monitoring of anti-Xa levels not usually necessary but anticoagulant effect can be measured by fondaparinux-specific anti-Xa assays.
Collection:
Fresh frozen plasma (FFP) is plasma that is prepared from a whole blood or apheresis collection and frozen at –18 C or colder within the time frame as specified in the manufacturer’s instructions for use of the blood collection, processing, and storage system (typically within 8 hours of phlebotomy):
- On average, units contain 200 to 250 mL, but apheresis-derived units may contain as much as 400 to 600 mL. FFP contains plasma proteins, including all coagulation factors.
- FFP contains normal levels of the labile coagulation factors, factors V and VIII.
- FFP should be infused immediately after thawing or stored at 1 to 6 C. After 24 hours, the component must be discarded or, if collected in a functionally closed system, may be relabeled as thawed plasma.
Recipient-donor compatibility:
Plasma must be ABO compatible with the recipient’s red cells. Compatibility with RhD is not necessary in plasma transfusion. Compatibility tests prior to transfusion are not necessary.
Indications:
FFP serves as a source of plasma proteins for patients who are deficient in or have defective plasma proteins.
Read more here.
Heparin is a negatively charged, sulfated glycosaminoglycan polysaccharide polymer isolated from porcine intestine, where it is stored in mast cell granules.
Learn more here.
Pain during dorsiflexion of foot; may be positive in patients with DVT. First described by John Homans in 1934. See image for description of test.
Humate-P is pasteurized plasma-derived von Willebrand factor (vWF)/factor VIII lyophilized concentrate with high vWF activity and higher proportion of high-molecular-weight vWF multimers than other vWF/factor VIII concentrates. Humate-P is FDA approved for adults and children with von Willebrand disease (vWD).
A humanized monoclonal antibody fragment targeted against the DOAC, dabigatran (idarucizumab binds to dabigatran with about 350 times greater affinity compared to thrombin). Indicated for the reversal of the anticoagulant effects of dabigatran in patients with life-threatening or uncontrolled bleeding or in patients having emergency surgery or urgent procedures.
More about idarucizumab:
- Half-life is about 45 minutes in patients with normal renal function
- Half-life is prolonged in patients with renal impairment
- Both thrombin-bound and free dabigatran are bound by idarucizumab
- Idarucizumab is given as a 5 g IV infusion (each dose given as 2 consecutive infusions of 2.5 g each)
- Adverse effects include
- Hypokalemia
- Delirium
- Constipation
- Fever
- Pneumonia
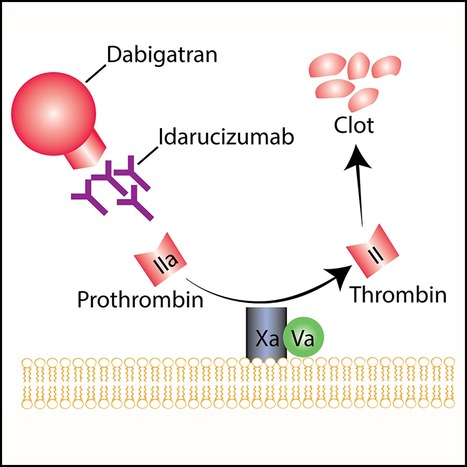
10 mg orally twice daily for 1 week
15 mg orally twice daily for 3 weeks
Purified from unfractionated heparin (UFH), LMWH has a more uniform polymer size vs. UFH, with a molecular weight of 3500 to 5000 daltons.
Learn more here.
Narrowing of left common iliac vein caused by compression/chronic pulsations of overlying right common iliac artery, leading to stasis of venous return within the lower extremity veins and deep vein thrombosis (DVT), most commonly of the left lower extremity. Originally described by May and Thurner in 1957. Learn more here.
Hemophilia A (90% of cases), hemophilia B (10% of cases)
The prothrombin time (PT)
Subtle hemostatic dysfunction without obvious clinical symptoms. May progress to overt DIC.
Decompensated hemostasis with bleeding and thrombotic manifestations.
Pathogen-reduced plasma is plasma that has been subjected to reduction technologies such as methylene blue, amotosalen or riboflavin treatment plus ultraviolet light, and solvent/detergent treatment to minimize transfusion-associated infections.
Pathogen reduced plasma products: a clinical practice scientific review from the AABB concluded in 2019:
The available evidence indicates that PR plasma products are as efficacious as conventional plasma products in a variety of patient populations and clinical conditions, except in the case of methylene blue and riboflavin PI…. With regard to non-infectious AE, this review has summarized evidence which, in aggregate, indicates that currently available S/D and PR plasma products have no documented deleterious effects compared to conventional plasma products. Furthermore, they appear to have lower rates of allergic reactions and TRALI.
Learn more here.
A fresh frozen plasma product in which the sample is placed:
- In refrigerator (at 1-6C) within 8 hours of collection.
- In freezer (at < -18C) within 24 hours of collection.
Venous congestion in deep vein thrombosis (DVT) impairing venous drainage, but sparing the superficial collateral vessels. As such, the arteries are not compressed and there is no tissue ischemia. Reported to precede phlegmasia cerulea dolens in 50%-60% of cases. Learn more here.
Complication of proximal deep vein thrombosis (DVT) caused by near-complete thrombotic occlusion of venous drainage, leading to severe venous congestion of lower limb. The congestion may, in turn compress the arterial circulation, leading to arterial insufficiency and ischemia. Patients may present with severe pain, swelling of the leg, cyanosis, and reduced or absent femoral pulse. Learn more here.
Persistent reduction of blood flow from leg resulting from incomplete resolution and/or healing of DVT. Symptoms may include chronic leg pain, swelling, redness, and less commonly ulcers.
Protamine sulfate is a small arginine-rich protein derived from salmon sperm nuclei. Forms a stable, inactive salt when bound to unfractionated heparin, rapidly reversing its anticoagulation effect. Onset of action within 5 minutes.
Purified concentrates of carboxylated vitamin K-dependent coagulation factors obtained from pooled human plasma and viral inactivation procedure.
HIT associated with abrupt platelet count fall (within 24 hours); occurs in patients who already have circulating anti-PF4/heparin antibodies because of recent heparin exposure (within the past 100 days).
Recombinant activated form of clotting factor, factor VII, originally developed to prevent or treat bleeding in patients with hemophilia A or B with inhibitors to factor VIII or IX, respectively. Available products include NovoSeven and Sevenfact.
- Functional assay that measures the ability of ristocetin to promote platelet aggregation in the presence of von Willebrand factor (vWF).
- Ristocetin is an antibiotic found to agglutinate platelets due to binding of vWF and platelet glycoprotein Ib (GPIb).
- Ristocetin binds to vWF, inducing a conformational change in vWF which increases its binding to platelets.
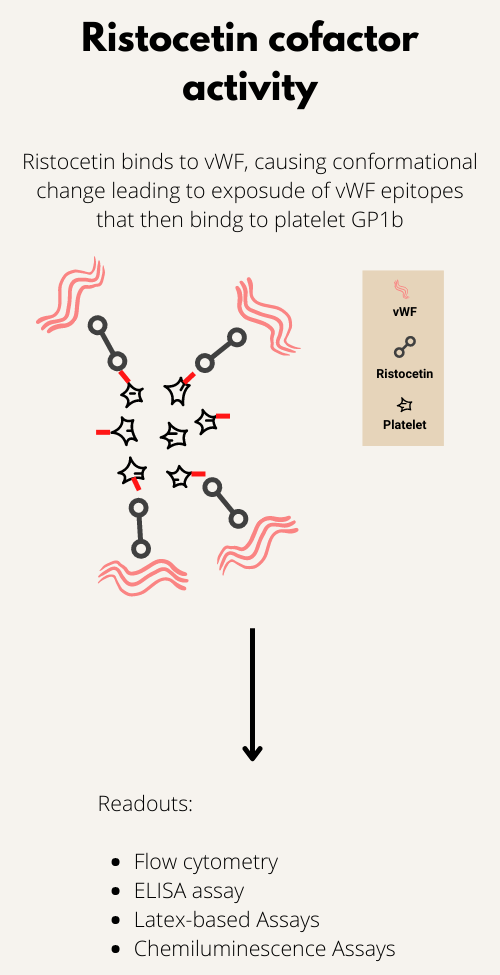
A rare variant of HIT that develops without prior heparin exposure.
Learn more here.
HIT in which platelets are recovered, but HIT antibodies are still positive.
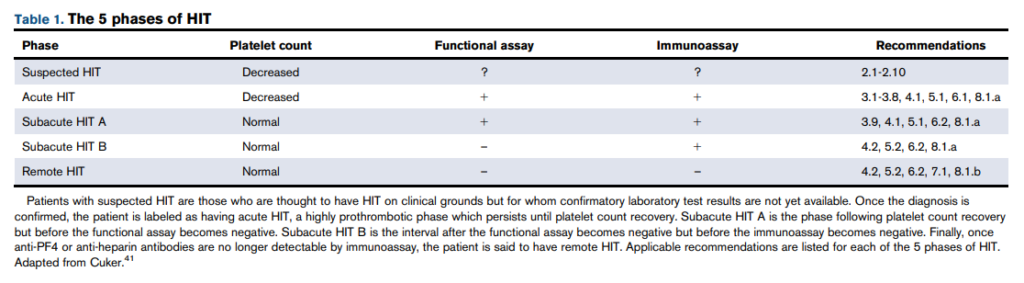
FFP, PF24, or PF24RT24 thawed and stored up to 5 days or original date of expiration (whichever is sooner) at 1-6 degrees C (33.8-42.8 degrees F).
A clinical prediction rule used to predict the likelihood (pretest probability) of HIT. Includes a consideration of magnitude of platelet count drop, timing of platelet count drop, presence of thrombosis and presence of alternative causes of the reduced platelet count. See calculator.
| Parameter | 2 points | 1 point | 0 points | Comment |
|---|---|---|---|---|
| Thrombocytopenia | Platelet count fall > 50% AND platelet nadir ≥ 20 × 109 L−1 | Platelet count fall 30%–50% OR platelet nadir 10–19 × 109 L−1 | Platelet count fall < 30% OR platelet nadir < 10 × 109 L−1 | Fall from highest platelet count that immediately precedes the putative HIT-related platelet count fall. 95% of cases of HIT are reported to develop in temporal association with heparin therapy; typically > 50% platelet count fall, but not to levels < 20 × 10 9 /L; only a few patients show 30%-50% platelet count fall; typical nadir is 40-80 × 109/L, with median of 55 × 109/L. |
| Timing of platelet count fall | Clear onset between days 5 and 10 OR platelet fall ≤ 1 day (prior heparin exposure within 30 days) | Consistent with days 5–10 fall, but not clear (e.g. missing platelet counts) OR onset after day 10 OR fall ≤ 1 day (prior heparin exposure 30–100 days ago) | Platelet count fall < 4 days without recent heparin exposure | Day 5 to 10 for initial platelet count fall with day 0 representing first heparin exposure; earlier fall if patient exposed to heparin with previous 30 days. Days are rounded off. For example, day 4.3 would count as day 4. |
| Thrombosis or other sequelae | New thrombosis (confirmed) OR skin necrosis at heparin injection sites OR acute systemic reaction after intravenous heparin bolus | Progressive or recurrent thrombosis or non-necrotizing (erythematous) skin lesions or suspected thrombosis (not proven. | None | |
| Other causes for thrombocytopenia | None apparent | Possible | Definite |
Scoring 0, 1, or 2 points for each of 4 categories, maximum possible score = 8 Low score (0-3 points), intermediate score (4 or 5 points), high score (6-8 points).
- Low score 0-3 points
- Intermediate score 4-5 points
- High score 6-8 points
Low 4Ts score may rule out HIT but high 4Ts score may not be sufficient to diagnose HIT.
1%-4%, with fatality rate of about 11%.
Used to predict VTE risk in surgical patients. Described by Caprini and colleagues in 1991. Score calculator can be found here.
Autoantibodies that recognize platelet factor 4 (PF4) bound to heparin; the resulting antibody-antigen complex activates platelets, resulting in mild to moderate thrombocytopenia and thrombosis. Learn more here.
A series of enzyme activation events in plasma, culminating in the formation of an insoluble fibrin clot.
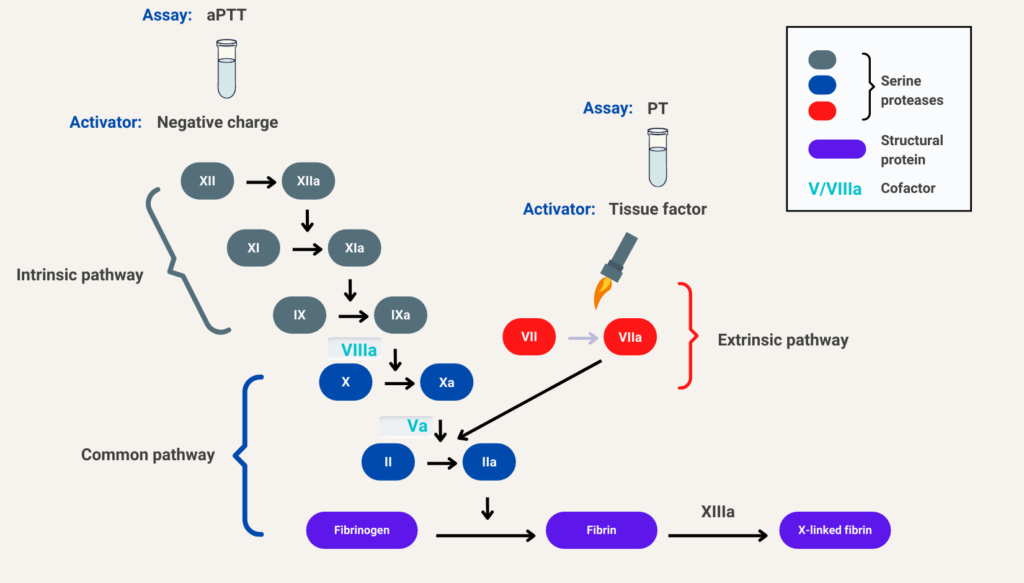
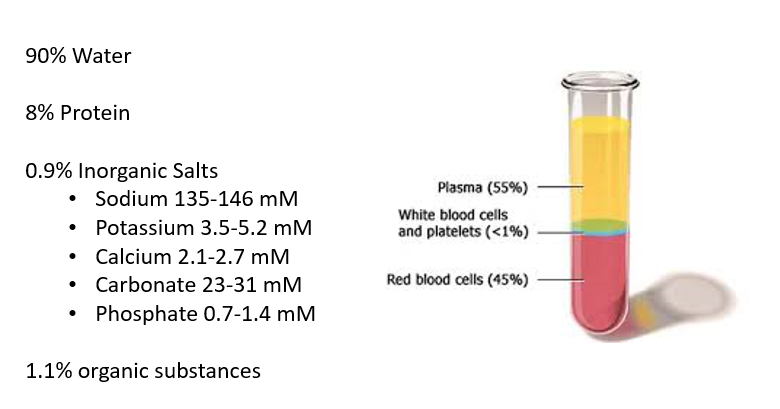
Defined by the Scientific and Standardization Committee (SSC) on DIC of the International Society on Thrombosis and Haemostasis (ISTH) as an acquired syndrome characterized by the intravascular activation of coagulation with a loss of localization arising from different causes; can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction.
3-factor PCC that contains low amounts of factor VII (FVII) and little/no protein C or protein S. 4-factor PCC that contains all vitamin K-dependent procoagulants and anticoagulants, including therapeutic amounts of FVII.
Warfarin is a generic name. Coumadin is a brand name for warfarin (hence, it is capitalized).
Activated PCC (aPCC) is composed mostly of nonactivated factors II, IX, and X, and activated factor VII (factor VIIa) as well as small amounts of proteins C and S, whereas PCC contains only inactive factors.
- Normal organ function → 2 μg/kg/min
- Liver dysfunction (bilirubin >1.5 mg/dL) → 0.5-1.2 μg/kg/min
- Heart failure, anasarca, postcardiac surgery → 0.5-1.2 μg/kg/min
Learn more here.
- Normal organ function → 0.15 mg/kg/h
- Renal or liver dysfunction → dose reduction may be appropriate.
- HITT: 15 mg twice per day × 3 weeks, then 20 mg once per day
- Isolated HIT: 15 mg twice per day until platelet count recovery

Learn more here.
Apixaban 10 mg orally twice daily for 1 week followed by 5 mg orally twice daily (reduce to 2.5 mg orally twice daily for extended use).
Dabigatran 150 mg orally twice daily after 5-10 day lead-in treatment with parenteral anticoagulation (unfractionated heparin or low-molecular-weight heparin [LMWH]).
Fondaparinux 5 mg subcutaneously once daily for patients with body weight < 50 kg; 7.5 mg subcutaneously once daily for patients with body weight 50-100 kg. Consider increasing dose to 10 mg subcutaneously once daily for patients with body weight > 100 kg.
Rivaroxaban 15 mg orally twice daily for 3 weeks followed by 20 mg orally once daily (reduce to 10 mg orally once daily for extended use.)
- Prevent recurrence.
- Prevent development of complications, including:
- Pulmonary embolism
- Post-thrombotic syndrome
- Minimize bleeding risk.
8-12 hours
14-17 hours
17-21 hours
3-6 hours; further increased in patients with renal insufficiency.
7-11 hours
About 1.5 hours
- 30 minutes after an IV bolus of 25 units/kg
- 60 minutes after an IV bolus of 100 units/kg
- 150 minutes after an IV bolus of 400 units/kg
About 36 hours
150 mg orally twice daily following ≥ 5 days of parenteral anticoagulation lead-in.
Suggested initial dosing is 5-10 mg orally to target INR 2-3. It is initiated with parenteral anticoagulation for ≥ 5 days and until INR is ≥ 2 for at least 24 hours, at which point parenteral anticoagulation can be discontinued. Learn more here.
The International Society on Thrombosis and Haemostasis (ISTH) scoring system was developed by the Scientific and Standardization Committee on Disseminated Intravascular Coagulation and published in 2001. It is makes use of laboratory tests available in almost all hospital laboratories.
| Parameter | Score | Comment |
|---|---|---|
| Fibrinogen (g/L) | >1 (0) <1 (1) | Overall sensitivity of low fibrinogen level reported to be about 30% |
| Prothrombin time (PT) (seconds) | <3 (0) 3-6 (+1) >6 (+2) | Elevated in 50%-60% of patients with DIC at some point during course of illness |
| Platelet count (109/L) | >100 (0) 50-100 (+1) <50 (+2) | Thrombocytopenia reported to occur in up to 98% of DIC cases |
| D-dimers or FDPs | No increase (0) Moderate increase (+2) Severe increase (+3) | Elevated FDPs and D-dimers are sensitive, but not specific for DIC |
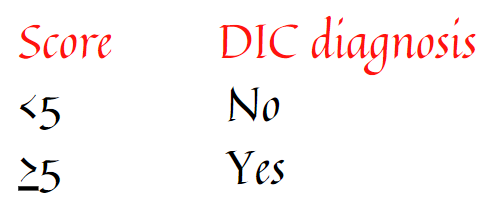
See ISTH calculator
Original publication on the ISTH score can be found here.
Coagulopathy, acidosis, and hypothermia. Learn more here.
Factor Xa inhibitor
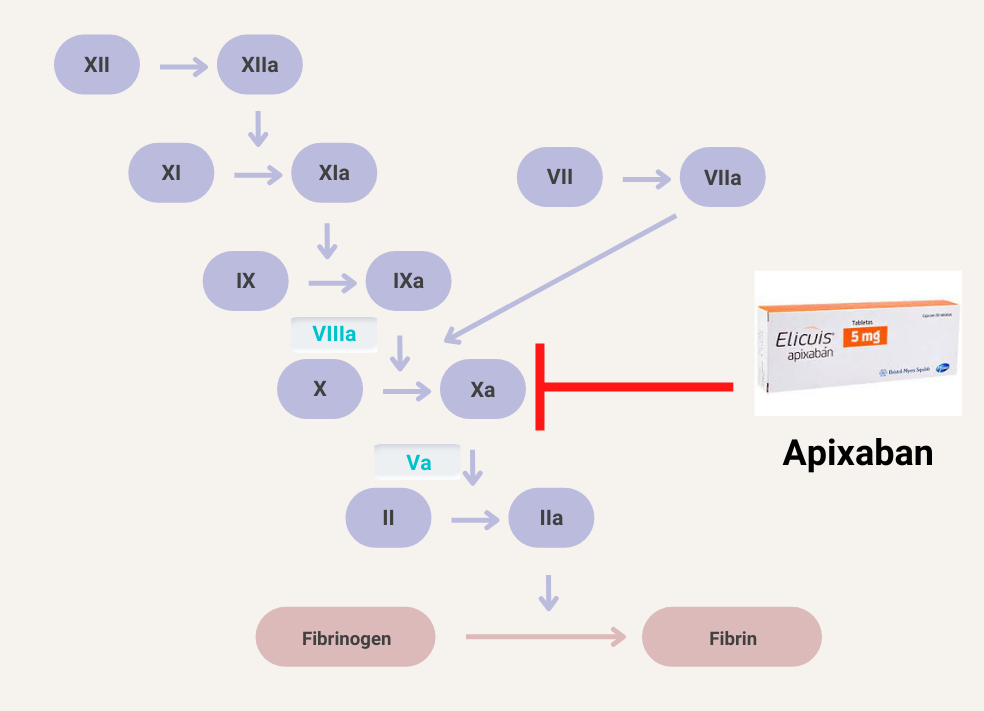
Thrombin inhibitor
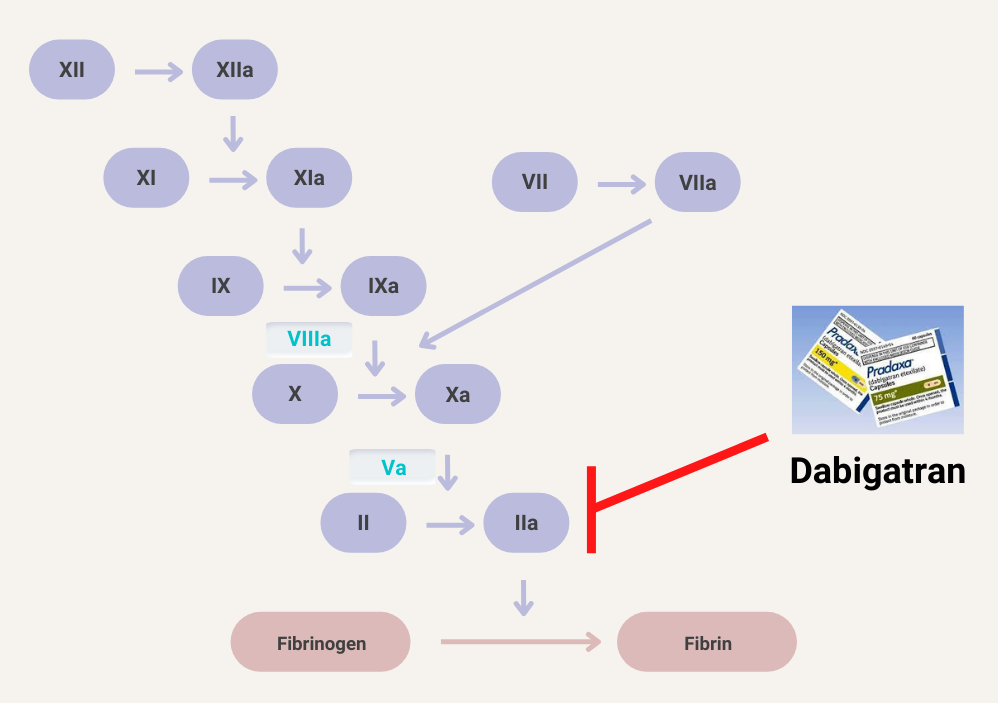
Factor Xa inhibitor
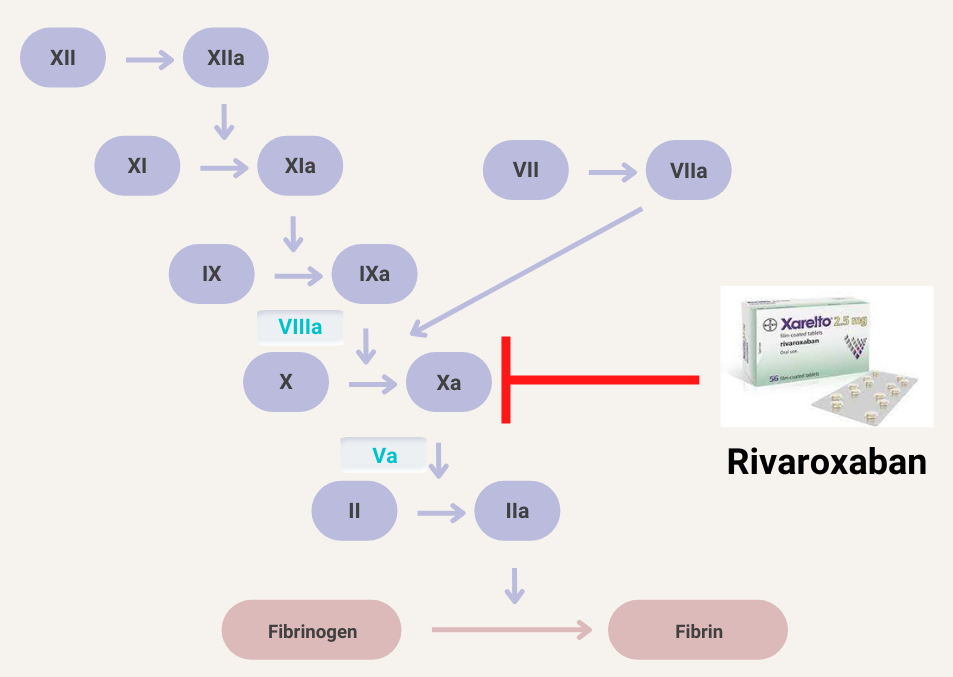

von Willebrand disease – prevalence 0.1-1% of general population
Veins, especially lower-limb deep vein thrombosis (DVT) and pulmonary embolism (PE).
Portal vein thrombosis
A rapid and reproducible test that measures primary platelet-related hemostasis using small amount of anticoagulated whole blood under conditions of high shear, with platelet plug formation. The assay is dependent on von Willebrand factor (vWF) and not fibrinogen.
Employs 3 cartridges, containing:
- Collagen (2 μg equine type I) and epinephrine (10 μg)-coated membrane
- Collagen (2 μg equine type I) and adenosine-diphosphate (50 μg)-coated membrane
- Prostaglandin E1 (5 ng) and ADP (20 μg)-coated membrane
For each cartridge:
- Whole blood (usually 800 μl) is placed into the sample reservoir, and the sample aspirated under a constant vacuum, passing with high shear force through a capillary and a microscopic aperture in the membrane.
- This results in platelet activation, attachment, and aggregation, forming a stable platelet plug at the aperture.
- The instruments report a ‘closure time’, which is the time required for full aperture occlusion and cessation of blood flow.
The instruments are very sensitive to von Willebrand factor defects/deficiencies, and therefore to VWD and to severe platelet function defects, including Bernard Soulier Syndrome or Glanzmann’s Thrombasthenia, but only moderately sensitive to mild platelet defect.
Both PFA-100 and PFA-200 systems employ similar mechanical processes, but the PFA-200 has more advanced software and a modern user interface including a touch screen.

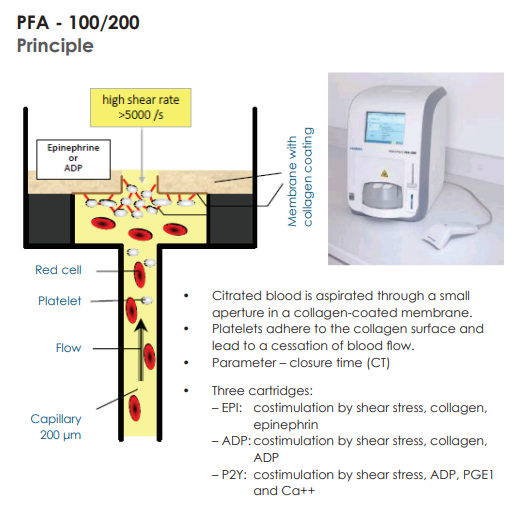
Learn more here.
About 15%, typically those with concomitant disseminated intravascular coagulation (DIC).
30-45% of patients
0.1% – 1% of the population.
UFH is associated with an ∼10-fold greater risk of HIT than LMWH.
Rivaroxaban, about 33%; Apixaban, about 25%; Dabigatran, about 80%.
About 80%
5% to 10% daily risk of thromboembolism, amputation, and death
Used to predict risk of postoperative VTE among non-cardiac surgery patients. The score was first described by Selwyn Rogers and colleagues in 2007. Score can be found here.
The SRA is a functional assay used to diagnose HIT. It identifies pathogenic IgG antibodies capable of binding and cross-linking platelet Fc gamma RIIA and triggering platelet activation. Test results are expressed as percentage of serotonin release compared to maximum release after detergent-induced platelet lysis (100%). Positive test is > 20% release at therapeutic heparin levels and < 20% release at supratherapeutic heparin levels. Specificity about 95%, but lower sensitivity reported compared with immunoassays.
1.5-2.5 x normal
Target INR 2.5 (therapeutic INR range 2-3)

- < 50 kg → 5 mg once per day
- 50-100 kg → 7.5 mg once per day
- >100 kg → 10 mg once per day
Eculizumab (plasma exchange within 24-48 hour of onset or admission if eculizumab is not immediately available). Eculizumab is a monoclonal C5 antibody that inhibits terminal complement complex formation.
40-80 × 109/L, with median of 55 × 109/L
5-10 days following first heparin exposure.
HIT is an iatrogenic disorder usually mediated by IgG antibodies that target multimolecular complexes of PF4 and heparin, leading to platelet activation, generation of procoagulant platelet-derived microparticles and activation of white blood cells and endothelial cells. The net result is a profoundly hypercoagulable state.
A clinical syndrome that includes microangiopathic hemolytic anemia, thrombocytopenia, and organ damage caused by endothelial cell injury and thrombotic occlusion of small blood vessels. Learn more here.
- The most common type; accounts for about 60%-85% of cases.
- The mildest form of disease.
- Autosomal dominant inheritance.
- Sequence variants absent in about 35% of patients.
- Plasma vWF level < 30 units/dL, parallel reduction in factor VIII levels.
- Variable degrees of bleeding.
- Bleeding in type 1 vWD is caused by a decrease in concentration of vWF, not a selective reduction in large multimers. All multimers are present in decreased amounts.
- Concordant reduction in VWF activity and antigen.
- About 15% of cases (called type 1C) are associated with increased clearance of VWF.
Learn more here.
Type 2 VWD is characterized by a qualitative abnormalities in the VWF protein. Different subtypes reflect which protein-protein interactions are affected. In some cases, reduced binding to a physiologic binding partner may be caused by defective multimerization rather than a defect in a specific protein binding domain:
- Accounts for about 20% of cases of vWD.
- Autosomal dominant inheritance.
- Subtypes include:
- Type 2A:
- Most common subtype.
- Accounts for 10 to 15 percent of VWD cases.
- Deficiency of large (intermediate and high-molecular-weight) multimers due to either:
- Decreased multimer assembly
- Increased proteolysis
- Impaired von Willebrand factor (vWF) binding to collagen and platelets.
- Type 2B:
- Accounts for approximately 5 percent of VWD cases.
- Gain-of-function mutation which causes increased vWF binding to platelet GPIb alpha with rapid clearance of platelet-vWF complex.
- Enhanced binding tom platelets leads to accelerated clearance or sequestration of platelets and of the bound HMW VWF multimers, resulting in deficiency of high-molecular-weight multimers and thrombocytopenia (the latter occurring in 40% of affected patients).
- Phenocopy of platelet type vWD.
- Type 2M (M for multimer):
- Loss-of-function mutation which decreases vWF binding to platelet GPIb alpha or collagen.
- Normal vWF multimer distribution.
- Type 2N (N for Normandy, the location where it was discovered):
- Loss-of-function mutations in vWF that cause decreased vWF binding to factor VIII with abnormally increased clearance of factor VIII.
- May mimic mild hemophilia A.
- Type 2A:
- Most cases caused by missense mutations, which are usually limited to specific functional domains.
- Ratio of von Willebrand factor ristocetin cofactor activity to von Willebrand factor antigen (vWF:RCo to vWF:Ag) typically < 0.7, with exception of type 2N and collagen-binding variant of type 2M.
Learn more here.
Type 3 vWD (autosomal inheritance) is the most severe form of vWD. It is associated with complete deficiency in vWF. Represents only 5% of all vWD. Learn more here.
2 pools of cryoprecipitate (5 units of cryoprecipitate/pool), which is expected to increase fibrinogen levels by 100 mg/dL (1 g/L).
Unfractionated heparin is a mixture of polymers with chain lengths ranging from 3000 to 30,000 daltons (mean, 15,000).
Learn more here.
Vascular wall damage or endothelial dysfunction, stasis of blood flow, and blood hypercoagulability
A bleeding disorder caused by quantitative or qualitative defects in von Willebrand factor (vWF) activity, which increases the risk of bleeding. Most cases are congenital (mutation in the VWF gene), though acquired vWD occurs in rare cases. vWD is the most common inherited bleeding disorder.
A multimeric plasma glycoprotein that mediates the adhesion of platelets to the subendothelial surface of blood vessels. Synthesized and released by endothelial cells and platelets, and circulates as large multimers of varying size.
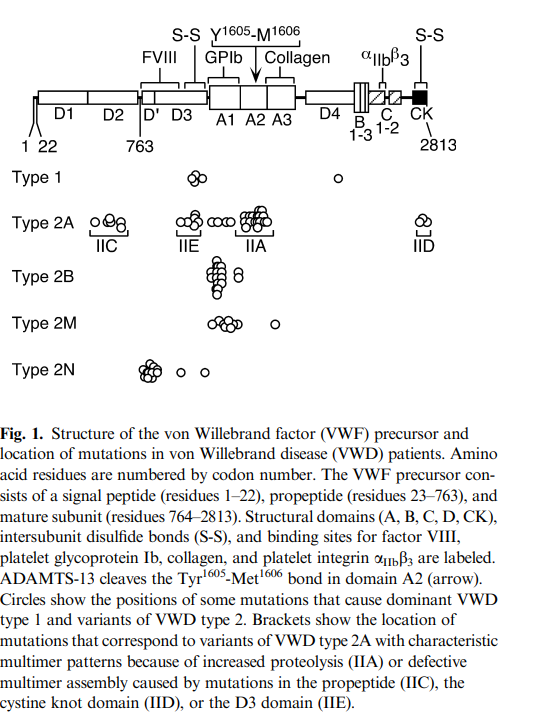
Vonvendi is a recombinant von Willebrand factor, FDA approved for use in adults with von Willebrand disease (vWD) for on-demand treatment and control of bleeding episodes. Contains large and ultra-large von Willebrand factor (vWF) multimers but lacks factor VIII (FVIII) so that patients may have to be supplemented with FVIII product.
Warfarin is the most common vitamin K antagonist (VKA) in clinical use:
- Racemic mixture of two optically active isomers, the R and S enantiomers.
- Highly water soluble.
- Rapidly absorbed from the GI tract.
- High bioavailability.
- Reaches maximal blood concentrations about 90 min after oral administration.
- Half-life of 36 to 42 h.
- Metabolized in liver by cytochrome enzymes.
Learn more here.
Wilate is a high-purity factor VIII/von Willebrand factor (vWF) concentrate FDA approved for adult and pediatric patients with von Willebrand disease (vWD) in whom desmopressin is ineffective or contraindicated for on-demand treatment and control of bleeding and perioperative management of bleeding.

Humate P, Alphanate, Wilfactin, Vonvendi
Prophylaxis for meningococcal infection (Neisseria meningitides) with vaccination, and antibiotics if treatment with eculizumab cannot be delayed until vaccine response. Per manufacture’s instructions:
- Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for meningococcal vaccination in patients with complement deficiencies.
- Immunize patients with meningococcal vaccines at least 2 weeks prior to administering the first dose of eculizumab, unless the risks of delaying eculizumab therapy outweigh the risk of developing a meningococcal infection.
- If urgent eculizumab therapy is indicated in an unvaccinated patient, administer meningococcal vaccine(s) as soon as possible and provide patients with two weeks of antibacterial drug prophylaxis.
Genetic abnormalities found in about 50%-70% of patients with atypical HUS. Learn more here.
It is estimated that plasma may contain as many as 40,000 different proteins from about 500 gene products. Approximately 1,000 proteins have been detected. 22 high-abundance proteins in the milligram per milliliter range account for 99% of the total protein content.

- Fresh frozen plasma
- Platelet transfusion
- Cryoprecipitate
- Fibrinogen concentrate
Stop the coumadin and reverse with vitamin K (at the same time stopping heparin and starting an alternative anticoagulant).

If INR 4.5-10 and patient not bleeding, decrease or withhold vitamin K antagonist (depending on INR level). If INR>10, and with no evidence of bleeding, oral vitamin K suggested (for example, 2.5-5 mg orally). For major bleeding, hospitalize patient, stop anticoagulant, administer vitamin K 5-10 mg IV and 4-factor prothrombin complex concentrate (PCC) (for INR 2-4, 25 units/kg, INR 4-6, 35 units/kg, and INR > 6, 50 units/kg).


Type 2B, as these patients are at risk for platelet aggregation and thrombocytopenia.
ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease:
“The panel suggests targeting both FVIII and VWF activity levels (trough) of >0.50 IU/mL for at least 3 days after surgery.”
Venous and arterial side of circulation (venous more common). Budd Chiari syndrome occurs in up to 25% of cases.
The VWF gene is located on the long arm of chromosome 12 and comprises 52 exons that encode 2813 amino acids.
Endothelial cells and megakaryocytes/platelets.
The peroneal, and the anterior and posterior tibial veins
Type 2B vWD (gain-of-function defect in VWF that causes enhanced VWF–platelet interactions via platelet GPIb).
- In type 2B VWD, VWF-GPIb platelet binding is pathologically enhanced, resulting in abnormal complexes between platelets and large adhesive forms of VWF. These complexes, not seen in other subtypes of VWD, are thought to account for the thrombocytopenia and depletion of large VWF multimers observed in many, but not all, cases of type 2B VWD.
- The degree of thrombocytopenia can vary and may be exacerbated at times of increased VWF production or secretion, such as during physical effort, inflammation, or pregnancy.
- Thrombocytopenia is rarely so severe as to be thought to contribute to clinical bleeding.
- Thrombocytopenia, while a distinctive feature of type 2B VWD, is not always present at baseline.
Learn more here.
Patients undergoing cardiac surgery
Erik Adolf von Willebrand was a Finnish internist who described the first case of vWD in 1926.

A Finnish physician, Erik von Willebrand, who published a Swedish-language description of a new bleeding disorder that he observed in a family living in the Aland Islands in the Baltic Sea in 1926.
From ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease:


Learn more here.
Platelets
Typically 5-10 days. Learn more here.
No. Normal complement concentrations do not exclude aHUS because low levels of circulating C3 have low sensitivity (about 30% of patients with aHUS), whereas high concentrations of circulating C5a and soluble C5b-9 might have insufficient specificity.
Learn more here.
Yes, especially in primary thrombocytosis, because platelet glycoprotein 1b binds to circulating von Willebrand factor, leading to acquired von Willebrand syndrome.
No, the pathogenesis of iron deficiency-associated thrombocytosis remains unclear, but may involve increased commitment of bipotential (erythroid/megakaryocytic) progenitor cells to the megakaryocytic lineage. Learn more here.
Yes, they are 20-25% lower in individuals with type O blood group. 14% of individuals with type O blood have vWF levels ≤ 50 units/dL. Learn more here.
According to the 2019 ASH guideline, <30 x 109/L:

According to the 2019 International Consensus Report, <20-30 x 109/L:

ADAMTS13 activity level
No. In unconfirmed aHUS, serum/plasma complement factor levels may suggest the diagnosis but they are not diagnostic.
Yes, and in fact they are now recommended as an option in the latest American Society of Hematology (ASH) guideline, especially for those who are not at high risk of bleeding and who do not have extensive clot burden:
At minimum, all critically ill non-bleeding patients with DIC should receive prophylactic anticoagulation for venous thromboembolism.
Regarding therapeutic doses of heparin, the 2013 ISTH harmonization guideline states:
Anticoagulant treatment may be a rational approach based on the notion that DIC is characterized by extensive activation of coagulation. Although experimental studies have shown that heparin can at least partly inhibit the activation of coagulation in DIC, there are no RCTs demonstrating that the use of heparin in patients with DIC results in an improvement in clinically relevant outcomes… therapeutic doses of heparin should be considered in cases of DIC where thrombosis predominates (low quality evidence). The use of low molecular weight heparin (LMWH) is preferred to the use of unfractionated heparin (UFH) in these cases (low quality evidence).
Yes! Proposed mechanisms include:
- Formation of platelet-white cell complexes
- Direct effect of pathogens on platelet activation and aggregation
- Hemophagocytosis
- Reduced platelet production
Learn more here.
Thrombocytopenia is absent at presentation in 15–20% of patients.
Learn more here.
Patients with type 2B vWD may have thrombocytopenia. Learn more here.
No, they are not different between the two conditions.
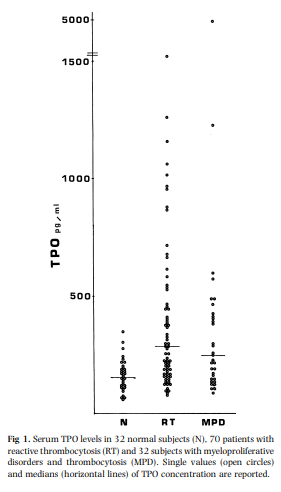
Learn more here.
Caplacizumab does not affect ADAMTS levels or ADAMTS13 antibody production. It is an anti–von Willebrand factor humanized, bivalent variable-domain-only immunoglobulin fragment that inhibits interaction between von Willebrand factor multimers and platelets.
Yes, it is much more common in later stages of cirrhosis (about 5% prevalence with I-IV fibrosis vs. 65% in patients with stage III-IV fibrosis). Learn more here.
CCI = posttransfusion count increment (CI; posttransfusion platelet count – pretransfusion platelet count) × body surface area (in m2)/number of platelets transfused
See calculator.
Posttransfusion platelet count – pretransfusion platelet count
By apheresis or from whole blood by platelet-rich plasma or buffy coat methods.
They are pinched off from proplatelet tips and released into the circulation. Learn more here.
They are stored in gas-permeable storage bags with gentle agitation at 20-24 degrees C (68-75.2 degrees F).
2-4 × 0.5 micrometers with mean volume of 7-11 fL.
15%-20% of patients presenting with acute lymphoblastic leukemia, > 90% of patients presenting with acute promyelocytic leukemia.
Over 95% of patients with DIC have thrombocytopenia.
Occurs in about 50-60% of such patients.
About 8%-68% of patients at time of admission, up to 50% of patients during stay in ICU. Learn more here.
Thrombocytosis occurs in about 10-15% of patients with iron deficiency anemia. Learn more here.
They block platelet aggregation by inhibiting fibrinogen binding to glycoprotein GPIIb/III on the platelet surface.
DDAVP 0.3 mcg/kg IV in 30-50 mL normal saline over 30 minutes. May be repeated 12-24 hours after first dose, depending on how well the patient responds, but no more than 2-3 doses should be given in order to avoid tachyphylaxis.
Steroids decrease platelet clearance and increase platelet production. Also, steroids may reduce bleeding, independent of their effect on platelet counts, by a direct stabilizing effect on blood vessels. Learn more here.
1 unit vWF:RCo/kg infused will result in recovery of 1.5-2 units vWF:RCo/dL in adults; 50 units vWF:RCo/kg will give a recovery of 75-100 units vWF:RCo/dL.
Direct toxic effect on the bone marrow. Learn more here.
Aspirin inhibits cyclooxygenase 1 (COX1), thereby decreasing production of thromboxane A, a major mediator of platelet activation.
DDAVP increases the release of vWF and factor VIII from endothelial cell stores by releasing vWF from cytoplasmic organelles called Weibel-Palade bodies.
Removes autoantibodies and replenishes ADAMTS13 levels.
Rarely below 30-40 × 109/L
How frequent are neurological symptoms in patients with thrombotic thrombocytopenic purpura (TTP)?
70%-80% of cases
Thrombosis occurs in one-third to one-half of patients with HIT. It may occur in veins, arteries or microvessels.
Platelet count < 100 × 109/L in absence of other causes of thrombocytopenia.

aPTT; adjust to 1.5-3.0 times baseline.
aPTT; adjust to 1.5-2.5 times baseline.
DDAVP may be administered IV, subcutaneously, or by nasal spray.
No one laboratory test is specific for DIC. Clinical practice guidelines recommend using a clinical scoring system, such as the ISTH scoring system, which includes consideration of the platelet count, D-dimers, fibrinogen and the prothrombin time.
Based on clinical presentation (thrombocytopenia and/or thrombosis in temporal association with heparin therapy without other obvious causes) plus presence of platelet-activating antiplatelet factor 4 (PF4)/heparin antibodies. Clinical prediction scores, especially the 4T score, are helpful in estimating the pretest probability of HIT and determining whether heparin should be discontinued and if further testing for antibodies should be carried out.

2019 Guidelines on the Use of Therapeutic Apheresis in Clinical Practice:
[Plasma exchange] is generally performed daily until the platelet count is >150 × 109/L, and LDH is near normal for 2-3 consecutive days. The role of tapering therapeutic plasma exchange (TPE) over longer duration has not previously been studied prospectively but is currently being reviewed. A small retrospective study suggests a lower overall recurrence rate at 6 months with taper. A common taper strategy is three times a week for the first week, twice weekly the second and then once weekly the following week(s). Other taper approaches have been documented.
Options include increased frequency of plasma exchange, rituximab, and immunosuppressive therapies such as cyclophosphamide and vincristine.
Primary (clonal) and secondary (reactive)
Platelet count > 450 × 109/L
ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease:
≤ 5 days in the United States; however, most blood collection centers culture apheresis platelets and release the unit 24-36 hours after collection.
4-8 weeks, including taper.
The 2019 ASH clinical guideline recommends treating for 6 weeks or less:

The 2019 International Consensus Report suggests a maximum of 8 weeks:

About 12 million in the United States!
About 1011 (by contrast, about 200 billion [200 x 109] red cells are released into the circulation every day)
1 apheresis platelet unit is equivalent to a pool of 4-6 units of whole blood-derived platelets; typically contains ≥ 3 × 1011 platelets.
About 5000.
Learn more here.
As of 2021, there are 27 known forms arising from mutations in 31 genes! Learn more here.
The current classification includes types 1 and 3, which are characterized by quantitative deficiencies of von Willebrand
factor (VWF), as well as types 2A, 2B, 2M, and 2N, which are qualitative variants.
Three main types are:
- Type 1 – partial quantitative deficiency of vWF.
- Type 2 – qualitative abnormalities of VWF; 4 subtypes:
- 2A – characterized by reduced or absent high-molecular-weight vWF.
- 2B – gain of function in VWF that increases its affinity for platelets.
- 2M – caused by reduced VWF interactions with platelets or collagen.
- 2N – results from reduced binding of VWF to FVIII.
- Type 3 – virtual absence of the VWF protein with associated very low FVIII levels.
10-20% of patients
If the patient has primary thrombocytosis (for example, essential thrombocythemia), they are at higher risk of thrombosis (though the risk does not correlate well with the platelet count). In patients with secondary or reactive thrombocytosis, there is no compelling evidence supporting an increased risk of thrombosis. Indeed, if anything, they may be at increased risk for bleeding owing to acquired von Willebrand syndrome (though this is far less common in secondary compared with primary thrombocytosis).
Once the platelet count is over 150 x 109/L, switch to warfarin (with appropriate overlap) or preferably to a DOAC (see 2018 American Society of Hematology [ASH] guideline recommendation 3.9 below). In case of HIT without thrombosis, continue for no longer than 3 months (typically 4-6 weeks). In the case of HIT complicated by thrombosis (termed HITT), treat for 3-6 months (see ASH guideline recommendation 3.8 below).
According to the 2019 International Consensus Report, may be repeated up to 3 times:

Even with platelet count recovery, patients remain at risk for thrombosis for 4 to 6 weeks after diagnosis (~17–53% over a thirty day period) because of circulating anti-PF4/heparin antibodies. While there is consensus among experts that patients with isolated HIT should be treated in the short-term with alternative anticoagulants, there is no firm consensus on the intensity or duration of anticoagulation for this patient population.
Platelet count recovery occurs within 7 days in 90% of cases. Functional assays become negative at a median of 50 days after heparin is suspended, while circulating anti-PF4/heparin antibodies are no longer detectable by immunoassay at a median of 85 days.
Median 50 days for platelet activation assays and 85 to 90 days for immunoassays.
No. For patients who have received heparin in the last 100 days, thrombocytopenia can develop rapidly (median = 10.5 hours), due to circulating PF4/H antibodies. However, patients who are re-exposed to the drug months to years after antibody disappearance do not manifest anamnestic responses, and seroconversion risk appears similar to de novo heparin exposure.
21-33 days postoperatively at median platelet count of 700 × 109/L before stabilizing after 45 days.
Day of first heparin exposure
Highest platelet count that immediately precedes the putative HIT-related fall in platelet count.
- < 72 hours post-surgery
- Infection (confirmed bacteremia/fungemia)
- Chemotherapy or radiation < 20 days
- Disseminated intravascular coagulation (DIC)
- Posttransfusion purpura
- Other drugs implicated in drug-induced thrombocytopenia
No. There are 4 parameters that are used in clinical scoring systems for DIC: fibrinogen, prothrombin time (PT), platelet count and D-dimers/fibrin degradation products. Fibrinogen is the least sensitive of these markers (overall sensitivity of low fibrinogen level reported to be 28%) because it is an acute phase reactant and therefore often elevated in conditions associated with DIC.
No, it is a complication of an underlying diseases such as sepsis or cancer.
Yes, in about 50% of cases. Learn more here.
No, there is no evidence that it is. Learn more here.
No. Platelet-associated IgG is elevated in both immune and nonimmune thrombocytopenia and assays for antibodies to specific platelet glycoproteins, while quite specific, have poor sensitivity. Clinical practice guidelines do not recommend ordering the test.

The aPTT may be increased in Type 2N vWD (the mutation specifically impairs the ability of vWF to carry FVIII) and Type 3 vWD (since vWF carries and stabilizes FVIII in the circulation, the very low levels of vWF associated with Type 3 vWD result in low FVIII activity).
There is no strong evidence for plasma therapy efficacy in atypical hemolytic uremic syndrome.
Learn more here.
No. Learn more here.
Yes, It’s called Vonvendi.
Yes, treatment with plasma exchange should begin as soon as possible, preferably within 4-8 hours of initial clinical diagnosis.


Not in patients with a typical presentation of ITP.


Antiplatelet therapy is not recommended, even in patients with extreme thrombosis, due to lack of evidence for thrombotic risk and theoretical risk of paradoxical bleeding from acquired von Willebrand syndrome. In cases where thrombosis has been reported in patients with reactive thrombocytosis, it is not known whether elevated platelet counts are causal or simply a marker of underlying disease with high thrombotic risk. Learn more here.
Yes, bilateral dopplers of the lower extremities are recommended in patients with acute isolated HIT.
Only if the patient has an upper-extremity central venous catheter, or has signs or symptoms suggestive of upper extremity DVT.
In a randomized trial, high-dose dexamethasone (40 mg daily x 4 days) was shown to lead to a faster response compared to prednisone, and without additional toxicity. However there was no difference in platelet count response at 6 months. Consider dexamethasone if quick response is particularly important, for example in a patient who is bleeding or needs to have surgery.
The following data are from the randomized trial mentioned above. The captions speak for themselves:
Guideline recommendations:


No, not unless there is another indication for these drugs, for example a coronary stent, and provided the risk: benefits are acceptable.
No, platelet transfusions may exacerbate the thrombotic tendency in HIT. Platelet transfusion should be reserved for patients with active bleeding or at high risk of bleeding.
Consider admission to hospital of patients with platelet < 20 x 109/L, even if they are asymptomatic.

No, platelet transfusions may exacerbate the thrombotic tendency in HIT. Platelet transfusion should be reserved for patients with active bleeding or at high risk of bleeding.
2018 American Society of Hematology (ASH) guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia
Consider the following comment from Good practice statements (GPS) for the clinical care of patients with thrombotic thrombocytopenic purpura:

It may be considered once the platelet count increases to a safe level (e.g., > 50 x 109/L), but there is no evidence of clinical efficacy in this setting.
From Good practice statements (GPS) for the clinical care of patients with thrombotic thrombocytopenic purpura:

- Reduce rates of alloimmunization.
- Reduce rates of febrile nonhemolytic transfusion.
- Prevent transmission of intracellular pathogens such as:
- Cytomegalovirus (CMV)
- Human T-lymphotropic virus
- Types I and II (HTLV- I/II)
- Immunoassays that detect presence of antibodies to platelet factor (anti-PF4/heparin antibodies).
- Functional assays – such as the serotonin release assay – that detect anti-PF4/heparin antibodies capable of activating platelets.
- Argatroban
- Bivalirudin
- Danaparoid
- Fondaparinux – especially in clinically stable patients at average risk of bleeding, and provided normal renal function.
- DOAC:
- Especially in clinically stable patients at average risk of bleeding.
- Most of the published experience in HIT is with rivaroxaban.
- Immune-mediated:
- Alloimmunization to human leukocyte, platelet-specific, or ABO antigens
- Autoimmune (immune thrombocytopenia)
- Non-immune-mediated:
- Hemorrhage
- Fever
- Sepsis
- Disseminated intravascular coagulation (DIC)
- Splenic sequestration
- Use of older stored platelets
- Thrombotic microangiopathy
Learn more here.
Essential thrombocythemia, polycythemia vera, primary myelofibrosis, CML, MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T), CMML and myelodysplastic syndrome with del (5q)
Aminocaproic acid and tranexamic acid
Quinine, quinidine, trimethoprim/sulfamethoxazole, and vancomycin (heparin-induced thrombocytopenia is considered separately).
- von Willebrand factor antigen (vWF:Ag)
- von Willebrand factor ristocetin cofactor activity (vWF:RCo) (platelet-binding activity of vWF measured by vWF:RCo assay)
- Factor VIII activity (FVIII:C)
- von Willebrand factor collagen binding (vWF:CB) activity (recommended by BCSH but not NHLBI as first-level test)
- Transfusion-associated graft-versus-host disease
- Immunocompromised patients
- Hematologic malignancies or certain solid tumors
- Recipients of marrow or peripheral blood stem cell transplantation
- Patients receiving blood components from blood relatives or human leukocyte antigen-compatible donors
- Patients receiving fludarabine therapy
- Prematurity, low birthweight, or erythroblastosis fetalis in newborn
Patients with low circulating platelet counts or functionally abnormal platelets, to prevent or control bleeding.
- Patients who are IgA deficient with documented presence of antibodies against IgA, for whom no IgA-deficient products are available.
- Patients experiencing posttransfusion purpura (washing can help remove complement).
- Patients with history of repeated severe allergic reactions to plasma-containing products.
- von Willebrand factor antigen (vWF:Ag)
- von Willebrand factor ristocetin cofactor (vWF:RCo) activity
- FVIII activity
Learn more here.
Thrombotic thrombocytopenic purpura (TTP) and heparin-induced thrombocytopenia (HIT)
- Unfractionated heparin > low molecular weight heparin (these differences are especially true at prophylactic doses)
- IV heparin > SC heparin
- Therapeutic or prophylactic dose heparin > heparin flush
- Bovine>porcine heparin
- von Willebrand factor (vWF) multimer assay
- Ristocetin-induced platelet aggregation (RIPA)
- vWF propeptide (vWFpp)
- vWF factor VIII binding (vWF:FVIIIB)
Platelets collected using automated instrumentation (also called apheresis platelets).
- Cyclooxygenase 1 (COX1) inhibitors such as aspirin
- P2Y12 inhibitors such as:
- Clopidogrel
- Prasugrel
- Ticagrelor
- Cangrelor
- Glycoprotein IIb/IIIa (GPIIb/IIIa) inhibitors such as:
- Tirofiban
- Eptifibatide
- Abciximab
- Thrombin receptor protease activated receptor inhibitor, vorapaxar
Systemic infection, solid and hematological malignancies, obstetric diseases (for example, abruptio placentae or amniotic embolism), trauma, aneurysms, and liver diseases.
- Neurological symptoms
- Pancreato-intestinal involvement
- Gangrene of the fingers or toes
- Ulcerative–necrotic skin lesions
- Myocardial infarction
- Ischemic cardiomyopathy
Learn more here.
abciximab, eptifibatide, and tirofiban
- Anemia (21%-39% of patients)
- Typically normocytic, normochromic)
- Rarely acquired hemolytic syndrome from hypophosphatemia accompanying anorexia nervosa (e.g., during the refeeding process)
- Leukopenia (29%-36% of patients)
- Mild neutropenia
- Lymphocytopenia
- Thrombocytopenia (5-11% of patients)
- Acanthocytes on peripheral smear
- Gelatinous transformation or serous atrophy on bone marrow exam
Learn more here.
Headache, facial flushing, tachycardia, and hyponatremia
Thrombotic type DIC and fibrinolytic type DIC.
Hereditary mutations in genes that encode thrombopoietin, the thrombopoietin receptor, or Jak2.
Another perspective:
World Health Organization requires 4 major criteria or first three major criteria + 1 minor criterion.
Major:
- Sustained platelet count > 450 × 109/L
- Bone marrow biopsy with characteristic changes
- Not meeting WHO criteria for BCR-ABL+ CML, PV, PMF, myelodysplastic syndromes, or other myeloid neoplasm
- Presence of JAK2, CALR, or MPL mutation
Minor: presence of clonal marker or absence of evidence for secondary thrombocytosis
Learn more here.
ISTH/SSC Harmonization of the recommendations:
- Fresh frozen plasma (FFP) recommended in patients with active bleeding or requiring an invasive procedure and either prolonged PT/aPTT (> 1.5 × normal) or decreased fibrinogen (< 1.5 g/L).
- Cryoprecipitate recommended for patients with active bleeding plus severe hypofibrinogenemia (< 1.5 g/L) that persists despite FFP replacement.
- Platelet transfusion recommended in patients with active bleeding plus platelet count < 50 × 109/L, or high risk of bleeding plus platelets < 20 × 109/L.
In patients with major bleeding (often in combination with corticosteroids) or in patients who require acute treatment and either cannot tolerate or do not respond to corticosteroids. Learn more here.


- Cyclooxygenase 1 (COX1) inhibitors such as aspirin
- P2Y12 inhibitors, including:
- Clopidogrel
- Prasugrel
- Ticagrelor
- Cangrelor
- Glycoprotein IIb/IIIa (GPIIb/IIIa) inhibitors:
- Tirofiban
- Eptifibatide
- Abciximab
Atypical HUS and infection-induced HUS, including Shiga toxin-producing Escherichia coli-associated (STEC) HUS and S. pneumoniae-associated HUS.
Systemic lupus erythematosus (SLE), infections (especially HIV and hepatitis C virus [HCV]), and lymphoproliferative disorders
Infection (with or without DIC) and medications (including heparin and non-heparin agents).
3 pillars of treatment are:
- Treat underlying disease that triggered DIC.
- Provide replacement therapy where appropriate.
- Inhibit thrombin and fibrin formation where appropriate.
- Type 1 – 75%
- Type 2 – 20%
- Type 3 – 5%
- Type 2A – decreased secretion or increased cleavage of high-molecular-weight multimers.
- Type 2B – increased vWF binding to platelet glycoprotein Ib (GPIb) alpha receptor with rapid clearance of platelet-vWF complex.
- Type 2M – defective binding to platelet GPIb alpha receptor or collagen; M for multimer.
- Type 2N – defective vWF binding to factor VIII; N for Normandy, where it was discovered.
Learn more here.
Increased platelet destruction, decreased platelet production and sequestration (hypersplenism).
A group of disorders characterized by microangiopathic hemolytic anemia, thrombocytopenia and microthrombi leading to ischemic tissue injury. Laboratory tests reveal thrombocytopenia and hemolytic anemia with red blood cell fragmentation (schistocytes) on the peripheral blood smear.
About 25% lower than in persons with other blood types.
Aging, pregnancy, inflammation (vWF is an acute phase reactant)
- Aging
- Pregnancy
- Stress (acute phase reactant)
- African Americans (vWF:Ag levels 20% higher compared to white patients, but vWF:RCo not affected)
Discontinue non-heparin anticoagulant and resume heparin.
- Continue heparin.
- No need for alternative anticoagulant.
- HIT antibody testing not recommended.
- Discontinue heparin.
- Initiate a non-heparin anticoagulant at therapeutic intensity.
- Test for HIT antibodies.
Anti-PF4/heparin antibodies of all isotypes (IgG, IgA, IgM, or polyclonal or polyspecific assays) or IgG isotypes.
Thrombocytopenia with normal Hb and white cell count.
A disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13
Atypical hemolytic uremic syndrome
1-desamino-8-D-arginine vasopressin, also called desmopressin
Disseminated intravascular coagulation
Drug-induced thrombocytopenia
Essential thrombocythemia
Heparin-induced thrombocytopenia
Heparin-induced thrombocytopenia with thrombosis
Immune thrombocytopenia
Myeloproliferative leukemia virus oncogene, also known as the thrombopoietin (TPO) receptor
Platelet factor 4
Shiga-like toxin producing Escherichia coli (E. coli) hemolytic-uremic syndrome (STEC-HUS)
Thrombotic microagniopathy
Thrombopoietin
Thrombotic thrombocytopenic purpura
von Willebrand disease
Von Willebrand factor
In one study:
- Normal platelet count in 84.6%
- Thrombocytosis (> 400 × 10 9/L) in 13.3%
- Thrombocytopenia (< 150 × 10 9/L) in 2.1%
Based on a landmark randomized study in 1991 (see abstract below), plasma exchange was reported to be associated with a mortality rate of about 10%, vs. 90% in historical controls.
- von Willebrand disease
- Inherited platelet function disorders (IPFDs), including:
- Bernard-Soulier syndrome
- Glanzmann thrombasthenia
- Montreal platelet syndrome
- Scott syndrome
- Dense granule storage pool disorders
- Alpha granule storage pool disorders
- MYH9-associated disorders
- Paris-Trousseau/Jacobsen syndrome
- Congenital amegakaryocytic thrombocytopenia
- Congenital malformation syndromes
Read more here.
Platelet count > 1,000-1,500 × 109/L
Most platelets are 1.5-3 μm in diameter. Small platelets are less than 1.5 μm in diameter. Large platelets usually range from 4 to 7 μm. Giant platelets are larger than 7 μm and usually 10-20 μm in diameter. Platelets that are larger than the size of the average red cell in the field qualify as giant platelets. Seen in many different reactive, neoplastic, and inherited conditions including myeloproliferative and myelodysplastic disorders, autoimmune thrombocytopenia, in association with severe leukemoid reactions, May-Hegglin anomaly and Bernard-Soulier syndrome.
150-450 x 109/L
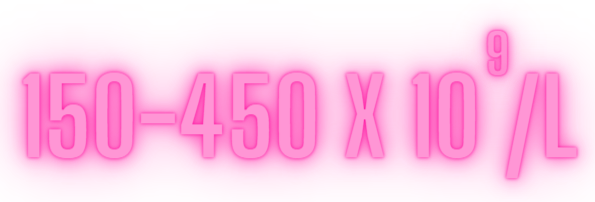
150-450 x 109/L
A platelet transfusion to prevent bleeding.
Alphanate is a high-purity factor VIII/von Willebrand factor (vWF) lyophilized concentrate used in patients with von Willebrand disease (vWD). it is FDA approved for adult and pediatric patients with vWD having surgery or invasive procedures in whom desmopressin (DDAVP) is ineffective or contraindicated.
Autosomal dominant disorder characterized by variable bleeding tendency, and variable association to hearing loss, cataracts, and nephritis. Encompasses Sebastian, May-Hegglin, Fechtner, and Epstein syndromes. Caused by mutation in MYH9 encoding nonmuscle myosin IIA. Learn more here.
Upshaw-Schulman syndrome
A type of thrombotic microangiopathy characterized by uncontrolled complement activation, microangiopathic hemolytic anemia, thrombocytopenia, and organ dysfunction, especially renal failure. Learn more here.
Autosomal recessive disorder characterized by moderate to severe bleeding, primarily mucocutaneous, from childhood. Characterized by mild thrombocytopenia with giant platelets, normal aggregation to all agonists except ristocetin and a prevalently severe bleeding phenotype. Learn more here.
A rare autosomal recessive disorder characterized by moderate to severe mucocutaneous bleeding, unusually large platelets, low platelet count (thrombocytopenia), and prolonged bleeding time (though this test is rarely performed). Caused by mutations in genes encoding proteins in the glycoprotein (GP) Ib/IX/V complex. Learn more here.
A humanized anti-von Willebrand factor (vWF) single-variable-domain immunoglobulin (nanobody) that targets AI domain of vWF, preventing its interaction with platelet glycoprotein 1b-IX-V, FDA approved for treatment of adults with immune TTP in combination with plasma exchange and immunosuppressive therapy.
HIT that develops or worsens after heparin has been discontinued. Thrombotic manifestations are delayed for days to weeks after heparin discontinuation and discharge.
According to Warkentin, TE:
There is a disorder called “delayed-onset HIT”. Despite its name, the timing of delayed-onset HIT resembles that of “typical onset HIT”, i.e. the platelet count fall begins 5–10 days after the immunizing exposure to heparin. Thus, there really is not any “delay” in onset of HIT (the term “delayed-onset” was intended to help the clinician remember that HIT can began several days after all heparin has been stopped). In recent years, the concept of delayed-onset HIT has expanded to include patients whose HIT worsens even after stopping heparin… patients’ HIT antibodies activate platelets in the absence of heparin, with activation inhibited by high heparin (100 IU/ml) and by Fcγ receptor blocking antibodies. These patients have a higher frequency of HIT-associated DIC, lower platelet count nadirs, and greater likelihood of severe sequelae, including venous limb gangrene. Sometimes, heparin “flushes” are the only proximate heparin exposure identified.
DDAVP is a synthetic derivative of antidiuretic hormone vasopressin that acts through type 2 vasopressin receptor.
Autosomal dominant disorder characterized by cleft palate, congenital cardiac abnormalities, developmental disabilities, facial dysmorphisms, immunodeficiency with parathyroid and thymus abnormalities, and mild to significant bleeding. Caused by microdeletion of chromosome 22q11.2 including the GP1BB gene (encodes glycoprotein 1b-beta [GPIbβ]). Learn more here.
Acquired clinicopathological syndrome characterized by systemic activation of coagulation that may cause organ-damaging thrombosis and/or hemorrhage due to consumption of coagulation factors and platelets. Defined by ISTH as “an acquired syndrome characterized by the intravascular activation of coagulation with a loss of localization arising from different causes; can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction”.
Eculizumab is an anti-C5 humanized monoclonal antibody that binds to C5, preventing its cleavage into C5a and C5b. Thus, eculizumab disables the terminal complement pathway and inhibits MAC-mediated intravascular hemolysis. It is first line therapy in atypical hemolytic uremic syndrome (aHUS) and paroxysmal nocturnal hemoglobinuria (PNH).
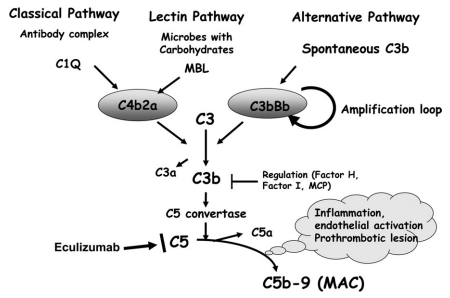
A vasomotor disturbance characterized by periodic attacks with triad of increased temperature, erythema, and burning pain resulting from hyperperfusion of skin areas, especially in the feet and hand. Typically relieved by cooling and aggravated by warming. May be primary or secondary. Causes of secondary erythromelalgia include polycythemia vera and essential thrombocythemia.
Learn more here.
Combined immune thrombocytopenia (ITP) and autoimmune hemolytic anemia. Learn more here.
A group of rare hereditary disorders associated with low platelet count and variable degree of bleeding complications. Learn more here.
Inborn error of metabolism leading to accumulation of glucocerebroside within lysosomes of cells.
Learn more here.
Autosomal recessive inherited platelet function disorder characterized by moderate to severe mucocutaneous bleeding. Caused by mutation in genes that encode for glycoproteins αIIbβ3 on platelet surface, leading to impaired platelet aggregation and increased risk of bleeding (especially mucocutaneous).
Humate-P is pasteurized plasma-derived von Willebrand factor (vWF)/factor VIII lyophilized concentrate with high vWF activity and higher proportion of high-molecular-weight vWF multimers than other vWF/factor VIII concentrates. Humate-P is FDA approved for adults and children with von Willebrand disease (vWD).
The prothrombin time (PT)
Thrombotic thrombocytopenic purpura (TTP), named after the physician Eli Moschcowitz who first described the condition.

Subtle hemostatic dysfunction without obvious clinical symptoms. May progress to overt DIC.
Decompensated hemostasis with bleeding and thrombotic manifestations.
Repeated failure to achieve adequate response to platelet transfusions
Genetic or acquired conditions associated with absent or reduced numbers of platelet granules or granules that are unable to empty their contents into the bloodstream. Learn more here.
Autosomal dominant condition associated with mild to moderate mucocutaneous bleeding caused by gain-of-function mutation in the megakaryocyte/platelet-specific gene encoding GPIbα, leading to increased affinity to von Willebrand factor, removal of high-molecular-weight von Willebrand factor from the circulation, and thrombocytopenia. Often confused with type 2B VWD, a disorder in which gain-of-function variants of VWF are responsible for increased GPIbα-vWF binding.
HIT associated with abrupt platelet count fall (within 24 hours); occurs in patients who already have circulating anti-PF4/heparin antibodies because of recent heparin exposure (within the past 100 days).
First-line therapy includes corticosteroids and/or IV immunoglobulin (IVIG [or anti-D immune globulin (anti-D)]. Patients who are not bleeding should be started on steroids (prednisone 1-2 mg/kg up to a maximum of 80 mg daily) or dexamethasone 40 mg PO or IV daily x 3 days. If there is a particularly high risk of bleeding, IVIG may be added to the regimen.
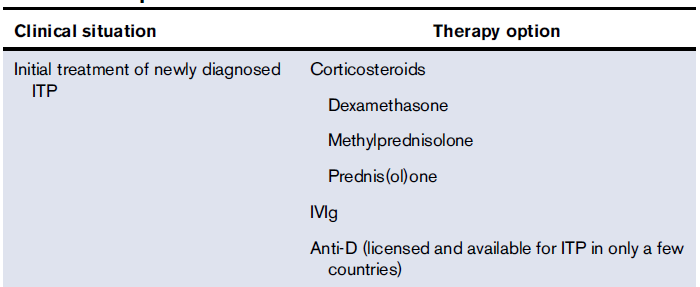

- Functional assay that measures the ability of ristocetin to promote platelet aggregation in the presence of von Willebrand factor (vWF).
- Ristocetin is an antibiotic found to agglutinate platelets due to binding of vWF and platelet glycoprotein Ib (GPIb).
- Ristocetin binds to vWF, inducing a conformational change in vWF which increases its binding to platelets.
Rituximab is a monoclonal antibody that targets CD20 antigen present on B lymphocytes.
Autosomal dominant or recessive disorder characterized by moderate to severe bleeding including menorrhagia, hematoma, and epistaxis. Caused by mutations in TMEM16F (ANO6) gene encoding transmembrane protein 16F (calcium-activated chloride channel). Learn more here.
A rare variant of HIT that develops without prior heparin exposure.
Learn more here.
- Thrombocytopenia that results from platelet clumping in vitro.
- An in vitro artifact with no clinical consequence.
- Usually caused by EDTA-dependent antibodies, but may also occur with poor specimen collection and/or handling.
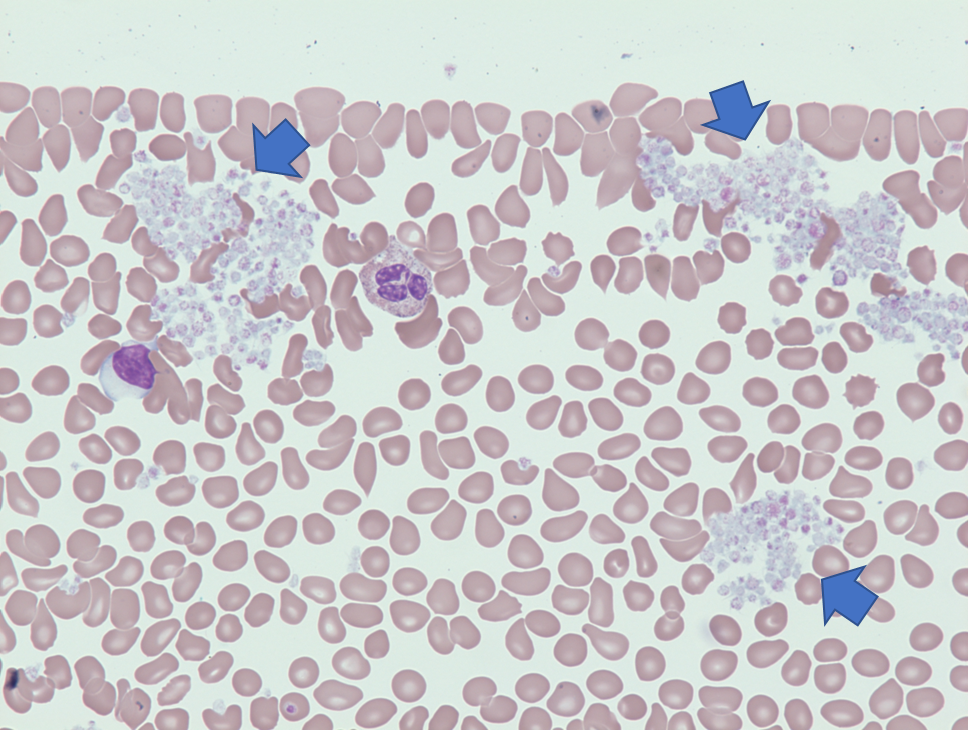
Hemolytic uremic syndrome caused by Shiga toxin-producing Escherichia coli (STEC).
Learn more here.
HIT in which platelets are recovered, but HIT antibodies are still positive.
A clinical prediction rule used to predict the likelihood (pretest probability) of HIT. Includes a consideration of magnitude of platelet count drop, timing of platelet count drop, presence of thrombosis and presence of alternative causes of the reduced platelet count. See calculator.
| Parameter | 2 points | 1 point | 0 points | Comment |
|---|---|---|---|---|
| Thrombocytopenia | Platelet count fall > 50% AND platelet nadir ≥ 20 × 109 L−1 | Platelet count fall 30%–50% OR platelet nadir 10–19 × 109 L−1 | Platelet count fall < 30% OR platelet nadir < 10 × 109 L−1 | Fall from highest platelet count that immediately precedes the putative HIT-related platelet count fall. 95% of cases of HIT are reported to develop in temporal association with heparin therapy; typically > 50% platelet count fall, but not to levels < 20 × 10 9 /L; only a few patients show 30%-50% platelet count fall; typical nadir is 40-80 × 109/L, with median of 55 × 109/L. |
| Timing of platelet count fall | Clear onset between days 5 and 10 OR platelet fall ≤ 1 day (prior heparin exposure within 30 days) | Consistent with days 5–10 fall, but not clear (e.g. missing platelet counts) OR onset after day 10 OR fall ≤ 1 day (prior heparin exposure 30–100 days ago) | Platelet count fall < 4 days without recent heparin exposure | Day 5 to 10 for initial platelet count fall with day 0 representing first heparin exposure; earlier fall if patient exposed to heparin with previous 30 days. Days are rounded off. For example, day 4.3 would count as day 4. |
| Thrombosis or other sequelae | New thrombosis (confirmed) OR skin necrosis at heparin injection sites OR acute systemic reaction after intravenous heparin bolus | Progressive or recurrent thrombosis or non-necrotizing (erythematous) skin lesions or suspected thrombosis (not proven. | None | |
| Other causes for thrombocytopenia | None apparent | Possible | Definite |
Scoring 0, 1, or 2 points for each of 4 categories, maximum possible score = 8 Low score (0-3 points), intermediate score (4 or 5 points), high score (6-8 points).
- Low score 0-3 points
- Intermediate score 4-5 points
- High score 6-8 points
Low 4Ts score may rule out HIT but high 4Ts score may not be sufficient to diagnose HIT.
Autoantibodies to ADAMTS13
Caused by mutations in genes encoding proteins in the megakaryocyte/platelet-specific glycoprotein (GP) Ib/IX/V complex.
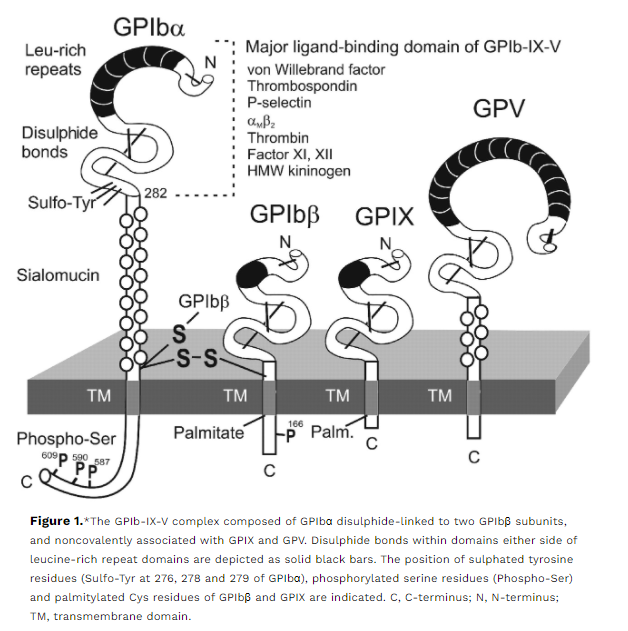
Quantitative and/or qualitative defect of glycoprotein αIIbβ3, resulting in the absence of platelet aggregation and increased risk of bleeding. Learn more here.
Autoantibodies that recognize platelet factor 4 (PF4) bound to heparin; the resulting antibody-antigen complex activates platelets, resulting in mild to moderate thrombocytopenia and thrombosis. Learn more here.
Congenital or acquired deficiency of the von Willebrand factor (vWF) cleaving protein, ADAMTS13
Autosomal recessive disorder characterized by variable oculocutaneous albinism, progressive neurologic dysfunction, severe immunodeficiency, recurrent life-threatening infections , and mild bleeding. caused by mutations in the LYST gene encoding a protein involved in granule trafficking and fusion.
- Thrombocytopenia,
- Microangiopathic hemolytic anaemia
- Damage to various organs, predominantly the kidney and the brain
Learn more here.
- Thrombocytopenia
- Microangiopathic hemolytic anemia (MAHA)
- Neurological changes (often fluctuating)
- Renal impairment
- Fever
The common feature to all forms of hemolytic uremic syndrome is the presence of endothelial cell lesions in the microvasculature of the kidney and, less frequently, of other organs… all forms of hemolytic uremic syndrome share a final common procoagulant and proinflammatory phenotype of activated endothelial cells.
Learn more here.
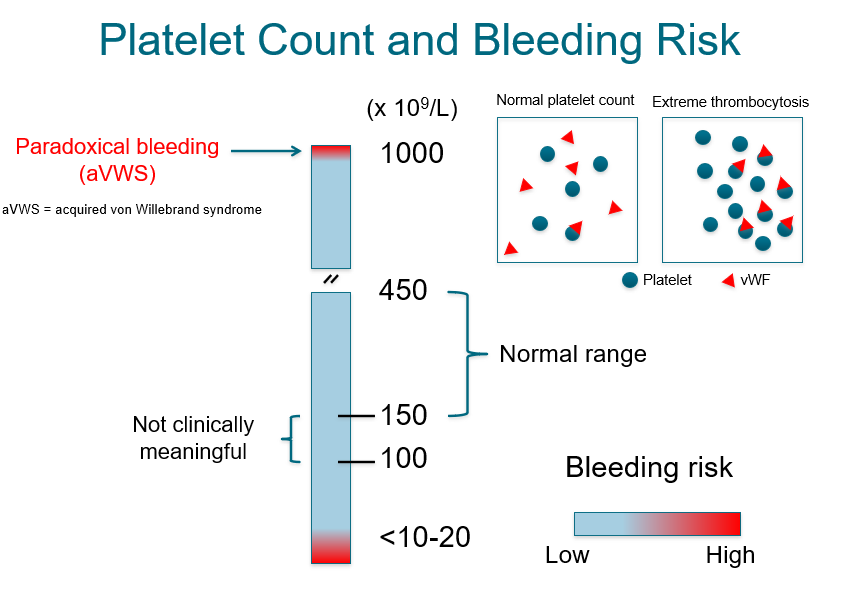
Defined by the Scientific and Standardization Committee (SSC) on DIC of the International Society on Thrombosis and Haemostasis (ISTH) as an acquired syndrome characterized by the intravascular activation of coagulation with a loss of localization arising from different causes; can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction.
Platelet count that is < the 2.5th percentile of normal platelet count range, with the traditional cutoff for the lower limit being 150 × 109/L.
Primary thrombocytosis is caused by a clonal disorder of hematopoietic stem cells or by inherited mutations (familial or congenital thrombocytosis). Secondary or reactive thrombocytosis occurs when signals (often cytokines like IL-11) promote proliferation and differentiation of normal megakaryocytes. Learn more here.
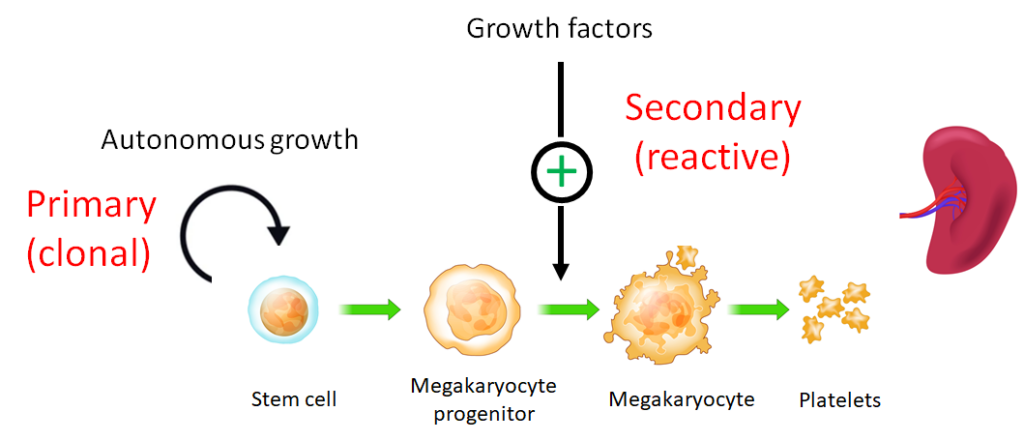
In primary TTP, there is no underlying predisposing condition. By contrast, secondary TTP is associated with another clinical diagnosis or underlying cause. In both cases, ADAMTS13 activity levels are, by definition, <10%.
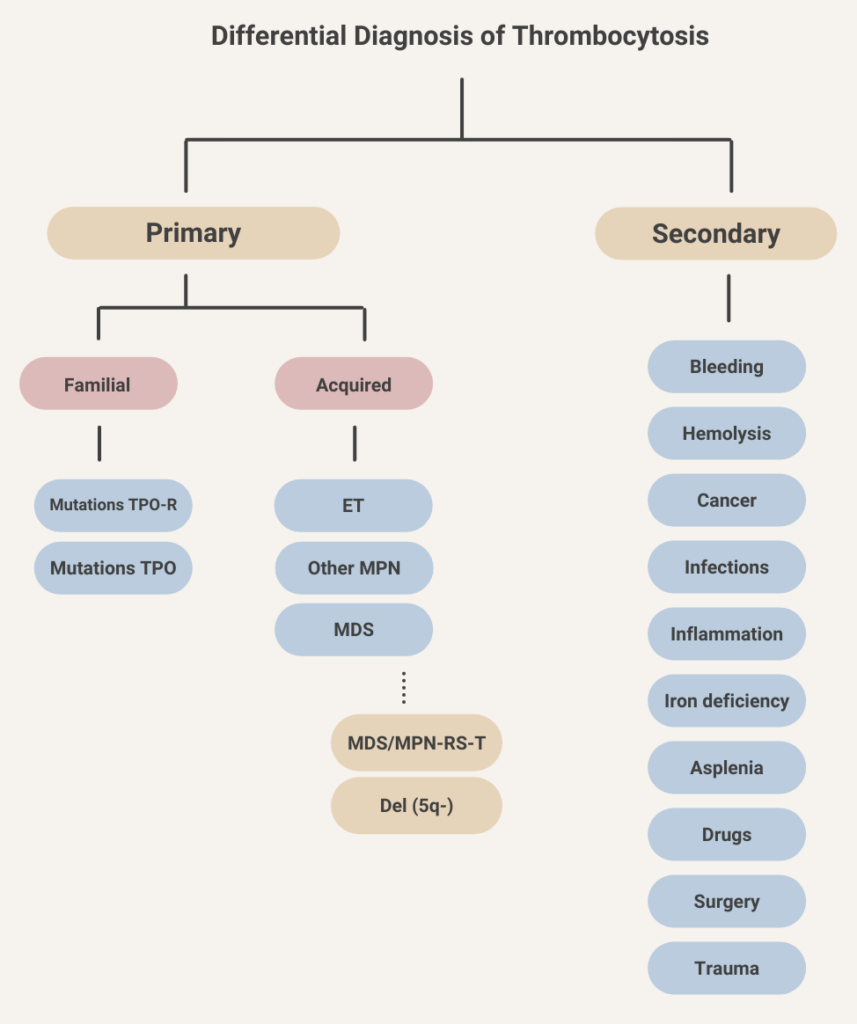
- Normal organ function → 2 μg/kg/min
- Liver dysfunction (bilirubin >1.5 mg/dL) → 0.5-1.2 μg/kg/min
- Heart failure, anasarca, postcardiac surgery → 0.5-1.2 μg/kg/min
Learn more here.
- Normal organ function → 0.15 mg/kg/h
- Renal or liver dysfunction → dose reduction may be appropriate.
375 mg/m2 IV weekly for 4 weeks.
- HITT: 15 mg twice per day × 3 weeks, then 20 mg once per day
- Isolated HIT: 15 mg twice per day until platelet count recovery
Learn more here.
Usually platelets > 30 x 109/L, though there are exceptions, e.g., in patients taking anticoagulants or anti-platelet agents, or in those undergoing a procedure.
Autosomal recessive disorder characterized by oculocutaneous albinism with nystagmus and visual acuity loss, immunodeficiency, mild bleeding pulmonary fibrosis, granulomatous colitis, and neutropenia, depending on the subtype. Caused by mutations in genes encoding proteins involved in vesicle formation and trafficking. Learn more here.
The International Society on Thrombosis and Haemostasis (ISTH) scoring system was developed by the Scientific and Standardization Committee on Disseminated Intravascular Coagulation and published in 2001. It is makes use of laboratory tests available in almost all hospital laboratories.
| Parameter | Score | Comment |
|---|---|---|
| Fibrinogen (g/L) | >1 (0) <1 (1) | Overall sensitivity of low fibrinogen level reported to be about 30% |
| Prothrombin time (PT) (seconds) | <3 (0) 3-6 (+1) >6 (+2) | Elevated in 50%-60% of patients with DIC at some point during course of illness |
| Platelet count (109/L) | >100 (0) 50-100 (+1) <50 (+2) | Thrombocytopenia reported to occur in up to 98% of DIC cases |
| D-dimers or FDPs | No increase (0) Moderate increase (+2) Severe increase (+3) | Elevated FDPs and D-dimers are sensitive, but not specific for DIC |
See ISTH calculator
Original publication on the ISTH score can be found here.
Meningococcal meningitis, which occurred in two of 100 patients included in prospective trials. Patients receiving eculizumab should receive specific meningococcal prophylaxis.
Learn more here.
7-10 days
Represents mean volume of platelets; usually reported as femtoliters (fL), calculated by dividing plateletcrit by number of platelets (analogous to calculation of mean red blood cell volume [MCV]). Normal range is 8-12 fL. It is high in cases of thrombocytopenia caused by peripheral destruction. MPV is low in cases of thrombocytopenia caused by megakaryocyte hypoplasia.
MPV is measured by most modern automated hematology analyzers, but the value is not always included in the complete blood count.
IVIG reduces Fc receptor-mediated clearance of platelets by the reticuloendothelial system.
8-10 g/dL
10-30 × 109/L
von Willebrand disease – prevalence 0.1-1% of general population
Veins, especially lower-limb deep vein thrombosis (DVT) and pulmonary embolism (PE).
Plasma exchange
Receptor for murine myeloproliferative leukemia virus, subsequently discovered to be the receptor for thrombopoietin, the primary driver of platelet production.
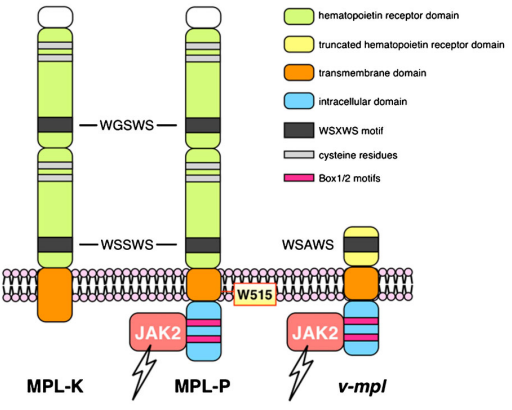
Learn more here.
Autosomal dominant disorder characterized by mild bleeding tendency, developmental delay, cardiac, skeletal, urogenital, central nervous system, gastrointestinal, and craniofacial abnormalities. Moderate to severe thrombocytopenia with giant alpha-granules in platelets that fail to release their contents upon stimulation with thrombin.
ADAMTS13 deficiency leads to reduced cleavage of ultra large vWF multimers, which leads to increased platelet aggregation and occlusion of small blood vessels.
A clinical prediction score that predicts severe ADAMTS13 deficiency in hospitalized patients with suspected thrombotic microangiopathy.

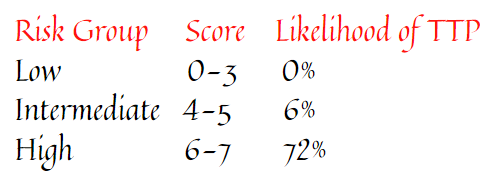
A rapid and reproducible test that measures primary platelet-related hemostasis using small amount of anticoagulated whole blood under conditions of high shear, with platelet plug formation. The assay is dependent on von Willebrand factor (vWF) and not fibrinogen.
Employs 3 cartridges, containing:
- Collagen (2 μg equine type I) and epinephrine (10 μg)-coated membrane
- Collagen (2 μg equine type I) and adenosine-diphosphate (50 μg)-coated membrane
- Prostaglandin E1 (5 ng) and ADP (20 μg)-coated membrane
For each cartridge:
- Whole blood (usually 800 μl) is placed into the sample reservoir, and the sample aspirated under a constant vacuum, passing with high shear force through a capillary and a microscopic aperture in the membrane.
- This results in platelet activation, attachment, and aggregation, forming a stable platelet plug at the aperture.
- The instruments report a ‘closure time’, which is the time required for full aperture occlusion and cessation of blood flow.
The instruments are very sensitive to von Willebrand factor defects/deficiencies, and therefore to VWD and to severe platelet function defects, including Bernard Soulier Syndrome or Glanzmann’s Thrombasthenia, but only moderately sensitive to mild platelet defect.
Both PFA-100 and PFA-200 systems employ similar mechanical processes, but the PFA-200 has more advanced software and a modern user interface including a touch screen.
Learn more here.
0·23–0·42 cases per million population. Learn more here.
About 1 in 5,000 hospitalized patients develop HIT.
About 15%, typically those with concomitant disseminated intravascular coagulation (DIC).
Thrombocytopenia reported in 77%-85% of patients with cirrhosis.
7%-10% of pregnancies are associated with thrombocytopenia. 70-80% of these patients have “gestational” thrombocytopenia.
0.1% – 1% of the population.
IVIG can be administered either high dose (1 g/kg daily for 1–2 days) in emergent settings or lower dose (e.g., 0.4 g/kg daily for up to 5 days). Learn more here.
Plasma exchange (mainstay of treatment; ideally started within 4-8 hours of initial clinical diagnosis) and methylprednisolone (1 g/day IV for 3 days for adults) or high-dose oral prednisolone (for example, 1 mg/kg/day) and (in some cases) rituximab or caplacizumab; for secondary TTP, same treatment as with primary TTP in addition to treatment of the underlying disorder.
Guideline recommendations:
ISTH guidelines for treatment of thrombotic thrombocytopenic purpura:



British Committee for Standards in Haematology (BCSH) guidelines on diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies:


UFH is associated with an ∼10-fold greater risk of HIT than LMWH.
5% to 10% daily risk of thromboembolism, amputation, and death
The SRA is a functional assay used to diagnose HIT. It identifies pathogenic IgG antibodies capable of binding and cross-linking platelet Fc gamma RIIA and triggering platelet activation. Test results are expressed as percentage of serotonin release compared to maximum release after detergent-induced platelet lysis (100%). Positive test is > 20% release at therapeutic heparin levels and < 20% release at supratherapeutic heparin levels. Specificity about 95%, but lower sensitivity reported compared with immunoassays.
Eculizumab, a monoclonal anti-C5 antibody that blocks the entry of C5 into C5 convertase.
Learn more here.
Eculizumab (plasma exchange within 24-48 hour of onset or admission if eculizumab is not immediately available). Eculizumab is a monoclonal C5 antibody that inhibits terminal complement complex formation.
40-80 × 109/L, with median of 55 × 109/L
5-10 days following first heparin exposure.
HIT is an iatrogenic disorder usually mediated by IgG antibodies that target multimolecular complexes of PF4 and heparin, leading to platelet activation, generation of procoagulant platelet-derived microparticles and activation of white blood cells and endothelial cells. The net result is a profoundly hypercoagulable state.
Congenital thrombotic thrombocytopenic purpura (TTP)


Learn more here.
X-linked recessive inheritance disorders characterized by eczema, microthrombocytopenia, immune deficiency, increased risk of developing lymphoma and autoimmunity, and clinically significant bleeding present from infancy. Wiskott Aldrich syndrome is caused by genetic changes in the WAS gene and is inherited in an X-linked manner. Wiskott-Aldrich syndrome, X-linked thrombocytopenia (XLT), and X-linked neutropenia (XLN) are known as ‘WAS-related disorders’ because these diseases are all caused by genetic changes in the WAS gene, and have overlapping symptoms ranging from severe to mild (Wiskott-Aldrich syndrome is the most severe).
A group of disorders characterized pathologically by occlusion of small (and occasionally large) blood vessels and clinically by thrombocytopenia, microangiopathic hemolytic anemia (MAHA) and end organ dysfunction. Learn more here.
A clinical syndrome that includes microangiopathic hemolytic anemia, thrombocytopenia, and organ damage caused by endothelial cell injury and thrombotic occlusion of small blood vessels. Learn more here.
A thrombotic microangiopathy (TMA) caused by deficiency of ADAMTS13, a von Willebrand factor cleaving protease, which leads to platelet-mediated occlusion of microvessels, thrombocytopenia, microangiopathic hemolytic anemia, and widespread multiorgan thrombosis and injury.
- The most common type; accounts for about 60%-85% of cases.
- The mildest form of disease.
- Autosomal dominant inheritance.
- Sequence variants absent in about 35% of patients.
- Plasma vWF level < 30 units/dL, parallel reduction in factor VIII levels.
- Variable degrees of bleeding.
- Bleeding in type 1 vWD is caused by a decrease in concentration of vWF, not a selective reduction in large multimers. All multimers are present in decreased amounts.
- Concordant reduction in VWF activity and antigen.
- About 15% of cases (called type 1C) are associated with increased clearance of VWF.
Learn more here.
Type 2 VWD is characterized by a qualitative abnormalities in the VWF protein. Different subtypes reflect which protein-protein interactions are affected. In some cases, reduced binding to a physiologic binding partner may be caused by defective multimerization rather than a defect in a specific protein binding domain:
- Accounts for about 20% of cases of vWD.
- Autosomal dominant inheritance.
- Subtypes include:
- Type 2A:
- Most common subtype.
- Accounts for 10 to 15 percent of VWD cases.
- Deficiency of large (intermediate and high-molecular-weight) multimers due to either:
- Decreased multimer assembly
- Increased proteolysis
- Impaired von Willebrand factor (vWF) binding to collagen and platelets.
- Type 2B:
- Accounts for approximately 5 percent of VWD cases.
- Gain-of-function mutation which causes increased vWF binding to platelet GPIb alpha with rapid clearance of platelet-vWF complex.
- Enhanced binding tom platelets leads to accelerated clearance or sequestration of platelets and of the bound HMW VWF multimers, resulting in deficiency of high-molecular-weight multimers and thrombocytopenia (the latter occurring in 40% of affected patients).
- Phenocopy of platelet type vWD.
- Type 2M (M for multimer):
- Loss-of-function mutation which decreases vWF binding to platelet GPIb alpha or collagen.
- Normal vWF multimer distribution.
- Type 2N (N for Normandy, the location where it was discovered):
- Loss-of-function mutations in vWF that cause decreased vWF binding to factor VIII with abnormally increased clearance of factor VIII.
- May mimic mild hemophilia A.
- Type 2A:
- Most cases caused by missense mutations, which are usually limited to specific functional domains.
- Ratio of von Willebrand factor ristocetin cofactor activity to von Willebrand factor antigen (vWF:RCo to vWF:Ag) typically < 0.7, with exception of type 2N and collagen-binding variant of type 2M.
Learn more here.
Type 3 vWD (autosomal inheritance) is the most severe form of vWD. It is associated with complete deficiency in vWF. Represents only 5% of all vWD. Learn more here.
A bleeding disorder caused by quantitative or qualitative defects in von Willebrand factor (vWF) activity, which increases the risk of bleeding. Most cases are congenital (mutation in the VWF gene), though acquired vWD occurs in rare cases. vWD is the most common inherited bleeding disorder.
A multimeric plasma glycoprotein that mediates the adhesion of platelets to the subendothelial surface of blood vessels. Synthesized and released by endothelial cells and platelets, and circulates as large multimers of varying size.
Vonvendi is a recombinant von Willebrand factor, FDA approved for use in adults with von Willebrand disease (vWD) for on-demand treatment and control of bleeding episodes. Contains large and ultra-large von Willebrand factor (vWF) multimers but lacks factor VIII (FVIII) so that patients may have to be supplemented with FVIII product.
Wilate is a high-purity factor VIII/von Willebrand factor (vWF) concentrate FDA approved for adult and pediatric patients with von Willebrand disease (vWD) in whom desmopressin is ineffective or contraindicated for on-demand treatment and control of bleeding and perioperative management of bleeding.
Humate P, Alphanate, Wilfactin, Vonvendi
Renal failure requires prompt initiation of dialysis in more than 50% of cases regardless of the cause of hemolytic uremic syndrome, and in more than 75% of adults with atypical hemolytic uremic syndrome.
Learn more here.
40–60% of patients.
Learn more here.
Platelet count < 10-20 × 109
Prophylaxis for meningococcal infection (Neisseria meningitides) with vaccination, and antibiotics if treatment with eculizumab cannot be delayed until vaccine response. Per manufacture’s instructions:
- Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for meningococcal vaccination in patients with complement deficiencies.
- Immunize patients with meningococcal vaccines at least 2 weeks prior to administering the first dose of eculizumab, unless the risks of delaying eculizumab therapy outweigh the risk of developing a meningococcal infection.
- If urgent eculizumab therapy is indicated in an unvaccinated patient, administer meningococcal vaccine(s) as soon as possible and provide patients with two weeks of antibacterial drug prophylaxis.
80% primary, 20% secondary
Genetic abnormalities found in about 50%-70% of patients with atypical HUS. Learn more here.
- Fresh frozen plasma
- Platelet transfusion
- Cryoprecipitate
- Fibrinogen concentrate
Stop the coumadin and reverse with vitamin K (at the same time stopping heparin and starting an alternative anticoagulant).
Type 2B, as these patients are at risk for platelet aggregation and thrombocytopenia.
Cardiac troponin, BUN and creatine, urinalysis, electrocardiogram and CT brain if neurological symptoms.

Mucocutaneous bleeding, including ecchymosis, petechiae, purpura, menorrhagia, epistaxis, and bleeding from lesions in the mouth and gastrointestinal tract.
Mucocutaneous bleeding is most common, including:
- Petechiae
- Purpura
- Ecchymoses
- Oral cavity bleeding
- Gastrointestinal bleeding
- Epistaxis
- Menorrhagia
- Hematuria
Intracerebral bleeding is rare. Hemarthrosis or extensive soft tissue hematoma suggest defects in secondary hemostasis.
Gray platelet syndrome, FI1B-RT, MYH9-related disease, and GATA1-related disease
WAS, XLT and CYCS-related thrombocytopenia
ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease:
“The panel suggests targeting both FVIII and VWF activity levels (trough) of >0.50 IU/mL for at least 3 days after surgery.”
Secondary (reactive) thrombocytosis accounts for ≥ 85% of cases.
The VWF gene is located on the long arm of chromosome 12 and comprises 52 exons that encode 2813 amino acids.
Endothelial cells and megakaryocytes/platelets.
Type 2B vWD (gain-of-function defect in VWF that causes enhanced VWF–platelet interactions via platelet GPIb).
- In type 2B VWD, VWF-GPIb platelet binding is pathologically enhanced, resulting in abnormal complexes between platelets and large adhesive forms of VWF. These complexes, not seen in other subtypes of VWD, are thought to account for the thrombocytopenia and depletion of large VWF multimers observed in many, but not all, cases of type 2B VWD.
- The degree of thrombocytopenia can vary and may be exacerbated at times of increased VWF production or secretion, such as during physical effort, inflammation, or pregnancy.
- Thrombocytopenia is rarely so severe as to be thought to contribute to clinical bleeding.
- Thrombocytopenia, while a distinctive feature of type 2B VWD, is not always present at baseline.
Learn more here.
Patients undergoing cardiac surgery
Robert S. Evans, first author on paper (titled “Primary thrombocytopenic purpura and acquired hemolytic anemia: evidence for a common etiology”) that first described the syndrome in 1951.
V. Koneti Rao wrote:
Known to his colleagues and friends as Bud Evans, he was a native Seattleite. Unfortunately, Evans was killed in a tragic car accident on Mercer Island in 1974 at the young age of 62.
Erik Adolf von Willebrand was a Finnish internist who described the first case of vWD in 1926.
A Finnish physician, Erik von Willebrand, who published a Swedish-language description of a new bleeding disorder that he observed in a family living in the Aland Islands in the Baltic Sea in 1926.
From ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease:
Learn more here.
Hemodilution and platelet consumption cause reduction in platelet count in early days post op (typically within 4 days of surgery), which leads to increased levels of thrombopoietin. The thrombopoietin response to acute thrombocytopenia takes 3 to 4 days to increase platelet production by bone marrow megakaryocytes, and results in a physiological “overshoot” in the platelet count. Postoperative platelet counts peak at two- to threefold the patient’s preoperative platelet count at approximately postoperative day 14, before gradually returning to the patient’s baseline value over the next 14 days. Learn more here.
White blood cells
Persons of African descent often have lower normal neutrophil counts, with ANC <1.5 × 109/L occurring in about 4.5% of black participants in one U.S. survey, and associated with the Duffy negative blood group. Learn more here.
Absolute neutrophil count (ANC) less than 1.5 × 109/L lasting for more than 3 months. Learn more here.
Constitutional/ethnic neutropenia is not associated with clinical manifestations. it is considered a normal variant (adaptive/protective in malaria-endemic regions of the world). It does not require any treatment, e.g., antimicrobial prophylaxis or G-CSF.
Lymphocyte count > 3.5-4.5 × 109/L
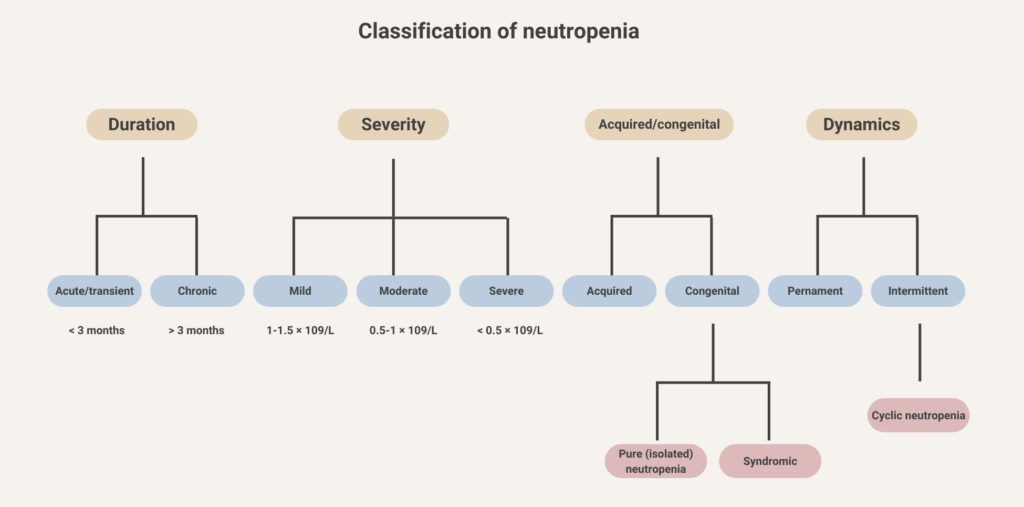
Absolute neutrophil count (ANC) ≤ 1.5 × 109/L in adults. Learn more here.
- Mild 1-1.5 × 109/L; does not impair host defense.
- Moderate 0.5-1 × 109/L; slight increased risk of infection if other arms of immune system also impaired.
- Severe < 0.5 × 109/L; increased risk of infection in most patients.
- Agranulocytosis ≤ 0.2 × 109/L; risk of severe, life-threatening infections.
Learn more here.
1.6 billion leukocytes per kilogram body weight; more than half are neutrophils.
- Arthralgia
- Bone and back pain
- Muscle spasms
- Musculoskeletal pain
- Pain in extremity
- Splenomegaly
- Epistaxis
- Chest pain
- Diarrhea
- Hypoesthesia
- Alopecia
- Familial or congenital (rare)
- Acquired
- Primary (clonal)
- Myeloproliferative neoplasms (MPN)
- Myelodysplastic syndrome (MDS)
- MDS/MPN overlap syndromes
- Acute myeloid leukemia (AML)
- Mastocytosis
- Myeloid/lymphoid neoplasms with eosinophilia (rearrangement of PDGFRA, PDGFRB, or FGFR1, or with PCM1-JAK2)Secondary (reactive)
- Secondary (reactive)
- Allergies
- Helminth infections
- Drug reactions
- Autoimmune disorders
- Cancer
- Lymphocyte variant hypereosinophilia syndrome (HES)
- Primary (clonal)
Learn more here.
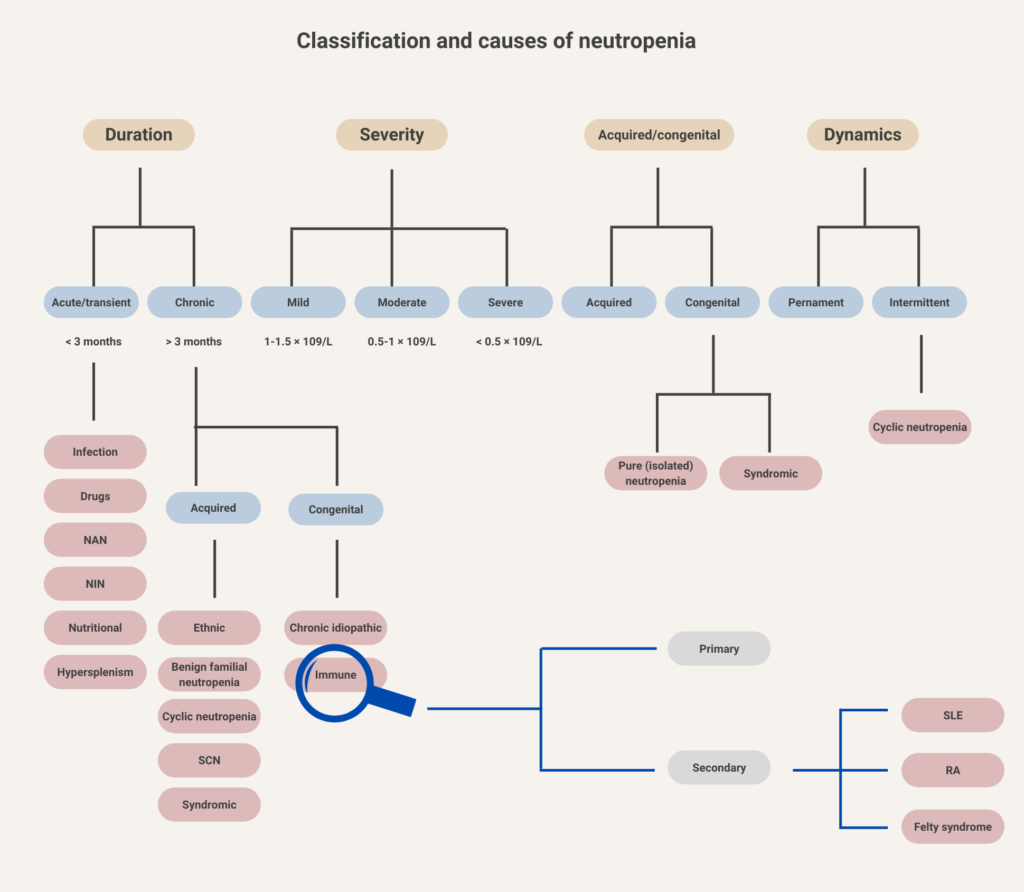

Neutrophils, basophils, and eosinophils
- Decreased production
- Increased retention in bone marrow (decreased release into blood)
- Increased migration to sites of inflammation
- Increased apoptosis
- Increased destruction and clearance by macrophages
- Splenic sequestration
A group of chronic myeloid disorders characterized by hematopoietic stem cell-derived clonal myeloproliferation.
Neutrophils, basophils, and eosinophils
- Anemia (21%-39% of patients)
- Typically normocytic, normochromic)
- Rarely acquired hemolytic syndrome from hypophosphatemia accompanying anorexia nervosa (e.g., during the refeeding process)
- Leukopenia (29%-36% of patients)
- Mild neutropenia
- Lymphocytopenia
- Thrombocytopenia (5-11% of patients)
- Acanthocytes on peripheral smear
- Gelatinous transformation or serous atrophy on bone marrow exam
Learn more here.

Another perspective:
- Chronic myeloid leukemia (CML) (BCR-ABL1+)
- Philadelphia chromosome-negative (BCR-ABL–) MPN:
- Polycythemia vera (PV)
- Primary myelofibrosis (PMF)
- PMF, prefibrotic/early stage
- PMF, overt fibrotic stage
- Essential thrombocythemia (ET)
- Chronic neutrophilic leukemia (CNL)
- Chronic eosinophilic leukemia, not otherwise specified (NOS)
- MPN, unclassifiable
Learn more here.
Döhle bodies bodies, toxic granulation and vacuolization
Reactive (secondary):
- Viral, bacterial, or parasitic infection
- Vaccinations
- Connective tissue disease
- Smoking
- Postsplenectomy state
Clonal (primary):
- Chronic lymphocytic leukemia (CLL)
- Certain B-cell and T-cell leukemia/lymphomas
Reactive (secondary):
- Viral or bacterial infection
- Corticosteroids
- Malignancy
- Postsplenectomy state
- Autoimmune disorders (such as inflammatory bowel disease or sarcoidosis)
- Vasculitides
Clonal (primary):
- Monocytic leukemia
- Chronic myelomonocytic leukemia
- Mastocytosis
- Infections
- Viral infections (most common cause)
- HIV
- Cytomegalovirus
- EBV
- HPV6
- Parainfluenza virus
- Parvovirus B19
- Bacterial infections, especially in cases of:
- Overwhelming infection (sepsis)
- Salmonella
- Brucellosis
- Malaria
- Viral infections (most common cause)
- Certain drugs – can occur at any time during treatment; most common culprits include:
- Chemotherapy agents
- Antiepileptics (e.g. valproate, carbamazepine)
- Antipsychotics (e.g. clozapine)
- Anti-thyroid drugs
- Antibiotics
- Anti-inflammatory drugs
Myelodysplastic syndrome
Myeloproliferative neoplasm
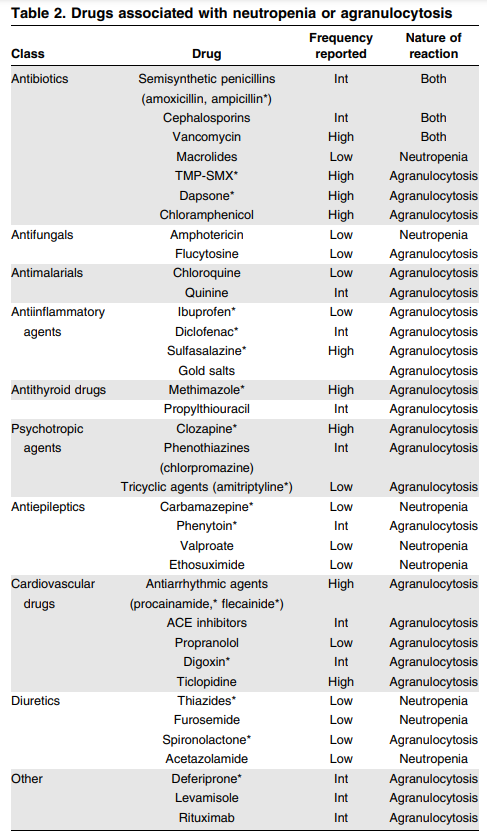
Differential counts of neutrophils, eosinophils, basophils, lymphocytes, and monocytes, typically performed by modern automated hematology analyzers.
A band neutrophil is a young neutrophil with deeply indented nucleus. The nucleus is indented to more that half the distance to the farthest nuclear margin. Chromatin is consistently present between edges of nuclear membrane, but in no area is it condensed to a single filament. Normally comprise < 10% of circulating white cells. Increased numbers in left shift (as occurs with inflammation and infection) and myeloproliferative neoplasms.
Döhle bodies appear as single or multiple blue or gray-blue inclusions of variable size (0.1 to 5.0 μm) and shape (round or elongated or crescent shaped) in the cytoplasm of neutrophils, bands, or metamyelocytes. They are often found in the periphery of the cytoplasm, near the cell membrane. Seen in conditions associated with increase cytokine release such as infection, burns, trauma, and administration of G-CSF.
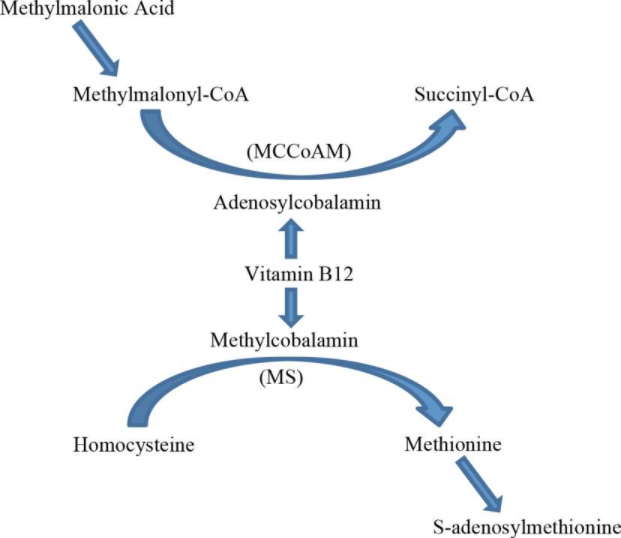
A leukemoid reaction is defined by a leukocyte count greater than 50,000 cells/μL, caused by reactive causes outside the bone marrow. Learn more here.
4-11 x 109/L
Smudge cells are remnants of fragile lymphocytes that are generated during preparation of peripheral smear. They lack any identifiable cytoplasmic membrane or nuclear structure. They contain dense nuclear material and or chromatic strands. They are characteristic of chronic lymphocytic leukemia.
Neutropenia that is acquired in adulthood but eludes a specific diagnosis is termed chronic idiopathic neutropenia (CIN). It is a diagnosis of exclusion that can only be made after a thorough and unrevealing search for other causes, including negative testing for autoimmune disease and nutritional deficiency and a normal bone marrow examination with normal cytogenetics. Its pathogenesis is unknown.
Learn more here.
Benign, stable course often in middle-aged women with rare evolution to myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). Diagnosis of exclusion. Term is often used synonymously with autoimmune-mediated neutropenia since there are no definitive tests to diagnose the latter.
Mild, chronic neutropenia, usually with an ANC >1000 in a patient with no history of recurrent infections. Affects mostly individuals of African or Arabic ancestry, but also seen in people from the Middle East or Crete. Learn more here.
Cyclic neutropenia (CN) is a rare autosomal dominant disease characterized by episodes of self-limited neutropenia that recur every 2 to 5 weeks. The cycles are of varying length but are very consistent within each patient. Unlike SCN, there is no increased risk of developing AML. CN results from mutations in ELANE.
Learn more here.
Persistent blood eosinophil count >1.5 x 109/L (on 2 examinations over ≥ 1 month without evidence of organ damage). Learn more here.
White blood cell count (WBC) > 100 x 109/L, usually seen in leukemias and myeloproliferative disorders, though may rarely occur as a reactive condition.
| Term | Definition |
|---|---|
| Leukocytosis | White blood cell (WBC) count > 11 × 109/L. |
| Leukemoid reaction | Increased WBC count (typically > 50-100 × 109/L) that occurs in response to reactive causes. |
| Hyperleukocytosis | WBC > 100 × 109/L, usually found in leukemias and myeloproliferative disorders. |
| Leukoerythroblastosis | Immature neutrophil precursors (with or without leukocytosis) and nucleated red blood cells in peripheral blood, indicative of severe disruption of the marrow by overwhelming infection, myelofibrosis, or bone marrow invasion due to malignancy. |
A benign condition diagnosed in young adults, with some resemblance to autoimmune neutropenia in children and the so-called ethnic neutropenia:
- Occurs in young adults.
- Because of the lack of any definitive tests, the diagnoses “autoimmune neutropenia” and “idiopathic neutropenia” are used nearly synonymously.
- Neutropenia may be mild, moderate or severe; may be associated with profound reduction in neutrophil count (< 0.5 x 109/L).
- Usually detected incidentally on a complete blood count.
- Those with severe neutropenia may develop recurrent fevers, upper respiratory infections and skin infections, but the infections are readily treated with antibiotics in most cases.
- The ANC may normalize during infections.
- Rarely associated with other organ-specific autoimmune diseases.
- Lasts several years or even life-long, with a female predominance (80% women).
Learn more here.
Immature neutrophil precursors – in absence or presence of elevated white count – and nucleated red blood cells (RBCs) in the peripheral blood, indicative of severe disruption of the marrow by overwhelming infection, myelofibrosis, or bone marrow invasion due to cancer. Tear drop shaped RBCs are also often present. Learn more here.
Intravascular accumulation of white blood cells (WBCs) causing obstruction of blood flow in patients with hyperleukocytosis (defined as WBC count >100 x 109/L), especially those with acute leukemia, in which the white cells are “stickier” compared to reactive causes or those with chronic lymphocytic leukemia (CLL). May lead to organ compromise. Learn more here.
A heterogeneous group of clonal disorders characterized by ineffective hematopoiesis leading to peripheral blood cytopenias and an increased risk of transformation to acute myeloid leukemia.
Reactive (polyclonal) eosinophilia secondary to malignancy
- A heterogeneous syndrome, caused by inherited mutations in several neutrophil-specific genes, most commonly the primary granule protein gene neutrophil elastase (ELANE).
- Characterized by agranulocytosis and recurrent, severe infections that begin during infancy.
- 10% to 30% lifetime risk of developing acute myelogenous leukemia.
learn more here.
Homozygosity of the single nucleotide polymorphism (SNP) rs2814778 located in the promoter region of the Duffy Antigen Receptor for Chemokines (DARC) also named Atypical chemokine receptor 1 (ACKR1) gene. This variant codes the Duffy negative [Fy(a-b-)] blood group. The Duffy positive phenotype is found in only 0.2% of Africans and 99.3% of Europeans. Duffy null polymorphism associated with protection against invasion of red blood cells by Plasmodium vivax malaria. Mechanism of lack of DARC and associated neutropenia unknown.
According to Donadieu et al:
Although this clinically insignificant variant most often seen in those of African descent has historically been called ‘benign ethnic neutropenia,’ we feel this name is problematic as it implies that ethnicity is causative and that this is a disease that requires intervention. We advocate for an alternative name such as chronic benign neutropenia, Duffy null associated neutrophil count, or typical neutrophil count with Fy (a-b-) status that reflects the genetic underpinning of this variant and highlights that it is not a disease state. Consensus has yet to be reached for an alternative name or expected ranges of normal neutrophil counts.
Autosomal dominant disorder usually caused by mutation in primary granule protein gene elastase, neutrophil expressed (ELANE, formerly ELA2), which encodes neutrophil elastase.
- Autosomal dominant causes include mutations in:
- ELANE
- GFI1, which encodes transcriptional repressor oncoprotein GFI1 thus repressing CEBPA, CEBPE and ELANE, important regulators of myeloid differentiation and neutrophil function
- CSF3R
- Autosomal recessive causes include mutations in:
- HAX1, which encodes an antiapoptotic gene (also called Kostmann syndrome)
- G6PC3, which encodes a neutrophil-specific catalytic subunit of glucose-6-phosphatase
- VPS45
- JAGN1
- CSF3R
- Unidentified genetic cause in up to 30% of SCN patients.
Absolute neutrophil count of 0.2 × 109/L or less
Basophil count > 0.1 x 109/L
White cell count > 11 x 109/L
Monocyte count > > 0.8-1.0 × 109/L
Neutrophil count > 7-7.7 × 109/L
Reactive (secondary):
- Drug reactions
- Allergy/hypersensitivity reaction
- Infections (especially tissue-invasive parasites, such as strongyloides)
- Cancer
- Sarcoidosis
- Connective tissue diseases
- Adrenal insufficiency
- Pulmonary diseases such as eosinophilia pneumonia
Clonal (primary):
- Chronic eosinophilic leukemia
- Myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1, or with PCM1-JAK2
- Other acute and chronic primary bone marrow processes such as myeloproliferative neoplasms
Reactive (secondary):
- Infection
- Chronic inflammation
- Smoking
- Stress
- Obesity
- Drugs, for example:
- Corticosteroids
- Beta-agonists
- Lithium
- Epinephrine
- Endocrine disorders:
- Hypercortisolism
- Thyroid storm
- Pre-eclampsia
- Post-splenectomy state
- Bone marrow stimulation
- Nonhematologic malignancy
- Inherited disorders
Clonal (primary):
- Chronic neutrophilic leukemia
- Chronic myelogenous leukemia
- Essential thrombocytosis (ET)
- Polycythemia vera (PV)
- Blood eosinophilia is defined as peripheral blood eosinophil count > 0.5 × 10 9/L.
- Blood hypereosinophilia is persistent eosinophil count > 1.5 × 109/L on 2 examinations over ≥ 1 month without evidence of organ damage.
- Hypereosinophilic syndrome is hypereosinophilia of any cause (reactive or clonal) associated with clear evidence of hypereosinophilia-related organ damage.
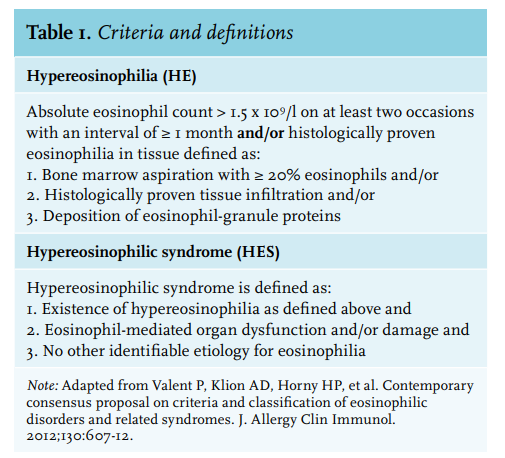
Learn more here.
- Defined as immature neutrophil count (myelocytes + metamyelocytes + promyelocytes)/total neutrophil count.
- Used as an indicator of a granulocyte left shift.
In adults, neutropenia is estimated to occur in about 1% of the non-African population and 5–8% of those of African
origin. Read more here.
Congenital neutropenia is extremely rare, with an estimated prevalence of 10-5. Learn more here.
Toxic granulation is the presence of large purple or dark blue cytoplasmic granules (primary granules) in neutrophils, bands, and metamyelocytes. The granules have increased staining density compared to normal neutrophils. Seen in conditions associated with increase cytokine release such as infection, burns, trauma, and G-CSF administration.
Only 1–5% of the total granulocytes in the body are circulating. Read more here.
Neutropenia reported to occur in up to 50% of patients with SLE.
Counts may vary considerably over short periods of time, associated with:
- Activity
- Exercise
- Eating
- Time
Learn more here.
Continue Exploring
Essays on Blood
Historical perspectives, fascinating blood tales in nature, and articles on blood
Learning Resources
Learn all things blood-related with Infographics, microcourses, quizzes and more
Blood for All
Discover the wonder of blood and what it means to have a hematological disorder