Heme biosynthesis and the porphyrias. The porphyrias are a group of at least eight metabolic disorders caused by mutations in the genes that encode enzymes involved in heme biosynthesis. Porphyrias differ considerably from each other, but a common feature in all porphyrias is the accumulation in the body of porphyrins or porphyrin precursors, depending on which specific enzyme of the heme biosynthetic pathway is affected. Heme precursors in the various porphyrias initially accumulate in either the liver or bone marrow, which are the tissues most active in heme biosynthesis. This is the basis for classification of porphyrias as either hepatic or erythropoietic. The major clinical manifestations of porphyrias are neurologic, usually presenting as acute attacks, or cutaneous, resulting from phototoxicity. Based on these differences, porphyrias are classified as acute or cutaneous. Enzyme activity is diminished in all but one of the porphyrias; in X-linked protoporphyria (XLP), increased activity is a consequence of gain of function mutations of the first enzyme of the pathway. Learn more here.
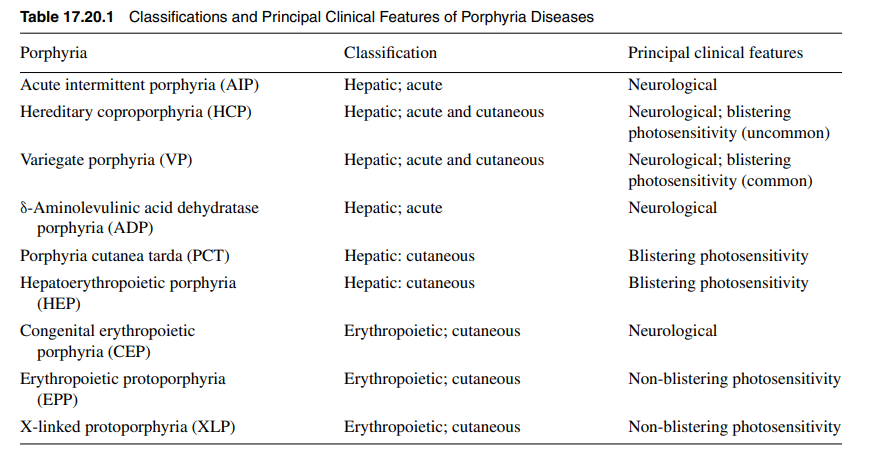
Another perspective of porphyria classification:
HEP, not shown in the graphic on the first slide, is a homozygous or compound heterozygous form of PCT, and results from a severe UROD deficiency. Only about 40 cases were reported up to 2010. Source