I posted a poll on twitter asking how you would treat a 23 yo F, 28 weeks pregnant, who presents to the ED with severe vaginal bleeding. There is no family history of bleeding. I showed a number of labs that included:
- An elevated aPTT, normal PT
- Low FVIII:C
- Normal von Willebrand factor activity
The choices for treatment included:
- FVIII concentrate
- vWF concentrate
- FVIII/vWF concentrate
- DDAVP
The most popular answer was FVIII concentrate:
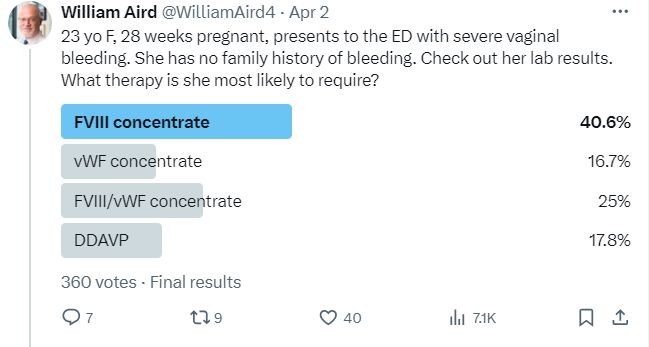
The correct answer was FVIII/vWF concentrate (DDAPV might work transiently in this case but given that her bleeding was severe, probably better to go with factor concentrates).
I made the case purposely vague to encourage you to think your way through it. Here is one possible way to navigate the information:
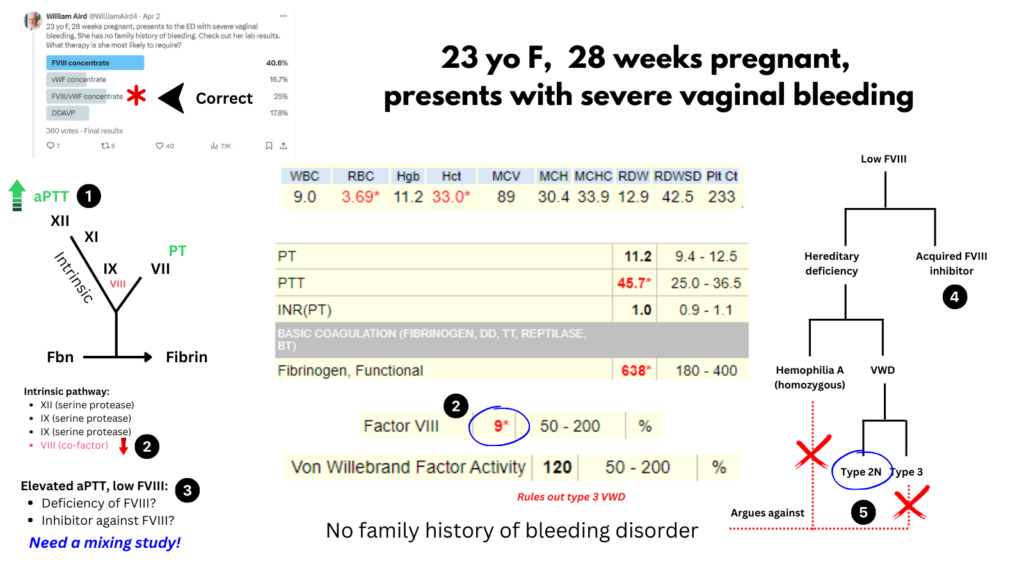
- Elevated aPTT and normal PT indicates a defect in the intrinsic pathway, which includes the serine proteases FXII, FXI, FIX and the cofactor FVIII (which accelerates FIX-mediated activation of FX).
- Factor FVIII activity (FVIII:C) is very low (9%) suggesting that it is responsible for the prolonged aPTT.
- Low FVIII may occur from a deficiency of FVIII or from an inhibitor (antibody) against FVIII. A mixing study would help sort this out, but we do not have one in this case.
- An acquired inhibitor against FVIII cannot be ruled out based on the information we are provided, and it has been rarely reported to occur in pregnancy, though usually in the postpartum period.1. However, treatment for an acquired inhibitor against FVIII (e.g., bypassing agent) was not an option in the poll.
- Hereditary deficiency of FVIII may occur in hemophilia A or VWD. Hemophilia A is X-linked, so this degree of FVIII deficiency in a woman would have to reflect homozygosity, which is not only extremely rare, but would present with a family history of hemophilia A in her father. There are two subtypes of VWD that are associated with this degree of FVIII deficiency: type 3 and type 2N. The normal VWF activity rules out type 3 VWD, leaving type 2N as the likely diagnosis. As we will see in this short review, type 2N VWD is treated with a combination of FVIII and VWF.
Description of the case:
- 23 yo F G2P0010 at 28w42 with known type 2N vWD presented with vaginal bleeding. She was diagnosed with type 2N VWD (using a VWF:FVIIIB binding assay) at age 11 after work up for unusual bleeding following dental procedure. She was treated with Humate-P prior to subsequent dental procedures. She had a trial of DDAVP 0.3 mcg/kg (18.8 mcg) with immediate post-infusion FVIII activity level increasing to 102.4. For this presentation, she was treated with Humate-P.
Bottom line
- Type 2N von Willebrand disease (VWD):2
- Qualitative defect in von Willebrand factor (VWF) caused by mutations that impair binding of VWF to coagulation factor VIII (FVIII), resulting in increased degradation and clearance of FVIII with reduced plasma FVIII levels.
- Clinical phenotype mimics an autosomal recessive form of hemophilia A.
- Diagnosis is:
- Suspected when FVIII:C levels are disproportionately low compared with VWF levels (FVIII:C/VWF:Ag ratio< 0.5).3
- Confirm diagnosis using assays for FVIII–VWF binding or DNA sequencing.
- Treatment of bleeding episodes may include:
- DDAVP (not always effective).
- Concentrates that contain both VWF and FVIII.
- N is for Normandy, where type 2N VWD was discovered.
Introduction
- Von Willebrand factor (VWF):
- A large, multimeric adhesive plasma glycoprotein.
- Synthesized by endothelial cells and megakaryocytes.
- Plays an important role in both primary and secondary hemostasis by:
- Mediating platelet adhesion/aggregation at sites of vascular injury.
- Acting as plasma carrier and stabilizer for FVIII.
- Von Willebrand disease (VWD):
- Bleeding disorder characterized by defects in VWF; VWD may be:
- Hereditary (more common) or acquired (rare)
- Qualitative or quantitative
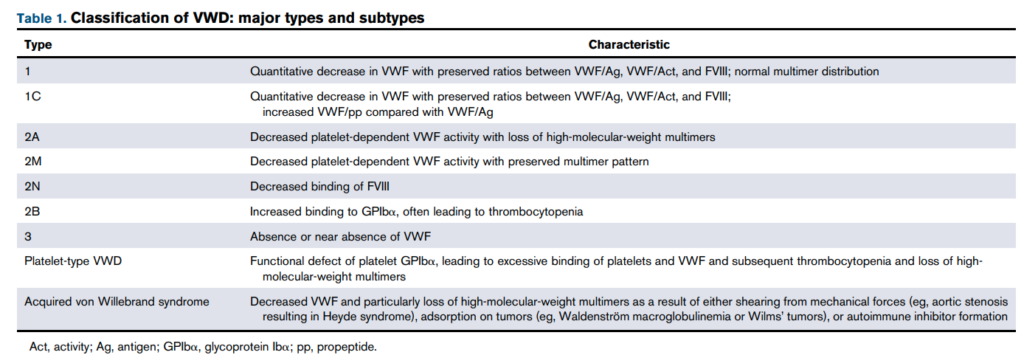
- Classified as:
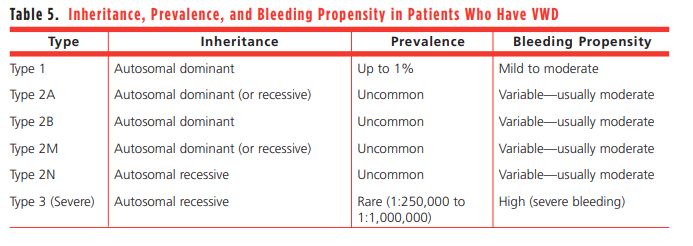
- VWD type 1:
- The mildest form of the disease.
- Associated with mild-to-moderate quantitative vWF deficiency (5-30 units/dL).
- Accounts for 60%-85% of cases and exhibits autosomal dominant inheritance.
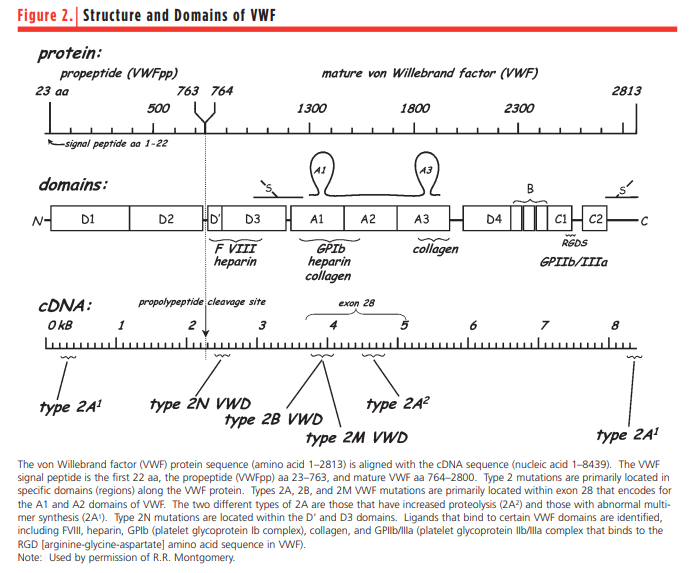
- VWD type 2:
- Comprises a qualitative defect in vWF, ranging from the absence of protein multimers to a defect in binding to platelets, collagen, or FVIII.
- Accounts for about 20% of cases and displays an autosomal dominant inheritance with the exception of type 2N, which is autosomal recessive.
- Type 2 is further subdivided on the basis of functional abnormalities:
- Type 2A:
- Most common subtype.
- Decreased secretion or increased cleavage of high-molecular-weight multimers, resulting in defective platelet-dependent VWF functions.
- Type 2B:
- Increased VWF binding to platelet glycoprotein Ib (GPIb) alpha receptor with rapid clearance of platelet-VWF complex, resulting in deficiency of large multimers (and occasionally thrombocytopenia).
- Type 2M
- Defective binding to platelet GPIb alpha receptor or collagen, resulting in defective platelet adhesion (normal multimer distribution).
- Type 2N:
- FVIII is produced normally.
- Defective VWF binding to FVIII, resulting in increased clearance of FVIII and a mild hemophilia phenotype (normal multimer distribution).
- Patients phenotypically resemble patients with hemophilia A.
- Type 2A:
- VWD type 3:
- The most severe type, caused by a complete deficiency in VWF, which may mimic hemophilia.
- Represents <5% of all cases and exhibits autosomal recessive inheritance.
- VWD type 1:
- Bleeding disorder characterized by defects in VWF; VWD may be:
History
- The importance of VWF for FVIII stability was first demonstrated by the observation that severe VWD (type 3 VWD) is associated with secondary FVIII deficiency.
- In 1989, Nishino et al reported 2 siblings from France with a new variant of VWD with a defective binding of VWF to FVIII, leading to lower levels of FVIII than VWF in plasma.
- In 1990, Mazurier et al reported the clinical and laboratory features of a 50-year-old woman from Normandy with a life-long history of excessive bleeding:
- Lab data showed FVIII deficiency and the presence normal plasma/platelet vWF multimers.
- The patient‘s vWF, in contrast to vWF purified from normal plasma, was unable to bind FVIII.
- The authors concluded: “These results support the concept that the bleeding diathesis of this patient appears to be due mainly to her abnormal vWF preventing FVIII/vWF interaction. This abnormality, which is not yet described in present classification of vWD, could be considered as a new variant of vWD… we tentatively suggest it be named not simply by a new letter but by the term “Normandy” after the patient’s birth province.
- In 1991, Tuley et al demonstrated a VWF missense mutation, Thr28 -* Met, in the patient originating from Normandy in or near the FVIII binding site. The corresponding mutant recombinant vWF (T28M) formed normal multimers and had normal ristocetin cofactor activity. However, vWF(T28M) exhibited the same defect in FVIII binding as natural VWF Normandy, confirming that this mutation causes the VWD Normandy phenotype.
Epidemiology
- Type 2N VWD represents:4
- 1% to 10% of all cases of vWD
- 10.6-13% of all type 2 VWD
Pathophysiology
- VWF is produced in endothelial cells and megakaryocytes, while FVIII is produced by endothelial cells, primarily in liver.
- FVIII circulates in plasma as a noncovalent complex with VWF, resulting in prolongation of its half life;5 approximately 95% to 98% of FVIII is in dynamic equilibrium complexed with VWF.67
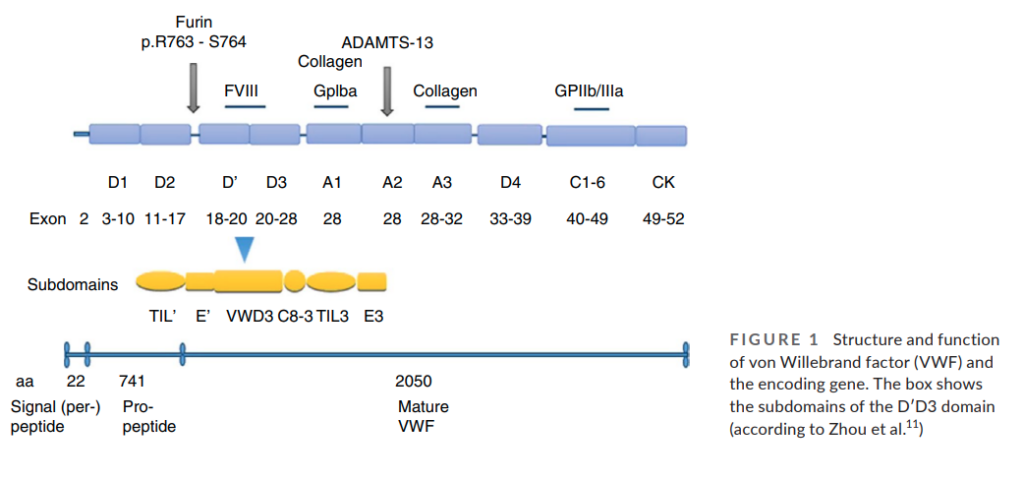
- The N-terminal D′D3 domain (encoded by exons 18–28 of VWF) is responsible for FVIII binding.8
- In type 2N VWD, mutations in the D′D3 domain result in:9
- Impaired binding of FVIII.
- Early proteolysis and loss of FVIII in the circulation.
- Reduced FVIII coagulant activity (FVIII:C) with normal or near-normal primary hemostasis and impaired secondary hemostasis.
- Other VWF functions are intact including binding to platelets and collagen.10
- More than 50 mutations have been described in type 2N VWD:11
- Most are located at the N-terminal of D′D3 (exons 18–20).
- The most common type 2N mutation, R854Q, is associated with a mild reduction of FVIII:C.
- Other mutations include C1060R, R816W, and T791M are associated with a more significant reduction in FVIII:C (<10%).
- In contrast to most cases of VWD, type 2N is inherited recessively and affected patients may be:12
- Homozygotes for a single 2N mutation
- Compound heterozygotes for two distinct 2N mutations
- Compound heterozygotes for a type 2N VWF mutation and a null VWF mutation.
Clinical presentation
- Mild to moderate hemophilia-like phenotype, including:1314
- Mucocutaneous bleeding (menorrhagia, epistaxis)
- Bleeding related to surgery or trauma
- Rarely hemarthroses and muscle hematomas
- Clinical expression depends on plasma levels of FVIII coagulant activity (FVIII:C).
- Those presenting with clinical manifestations include:15
- Homozygotes
- Compound heterozygotes for:
- Two different type 2N mutations
- A combination of 2N and another VWD type
- Heterozygotes are generally asymptomatic when they have normal or only mildly reduced FVIII:C.
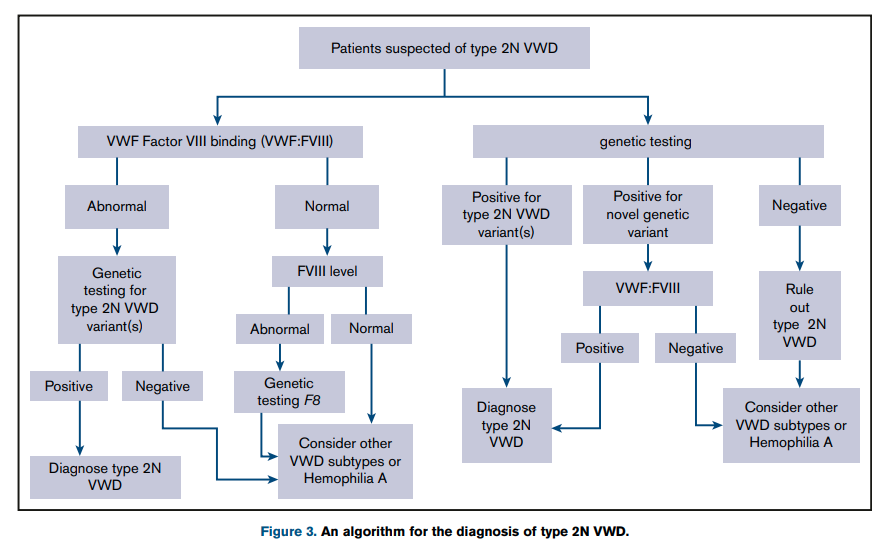
Diagnosis
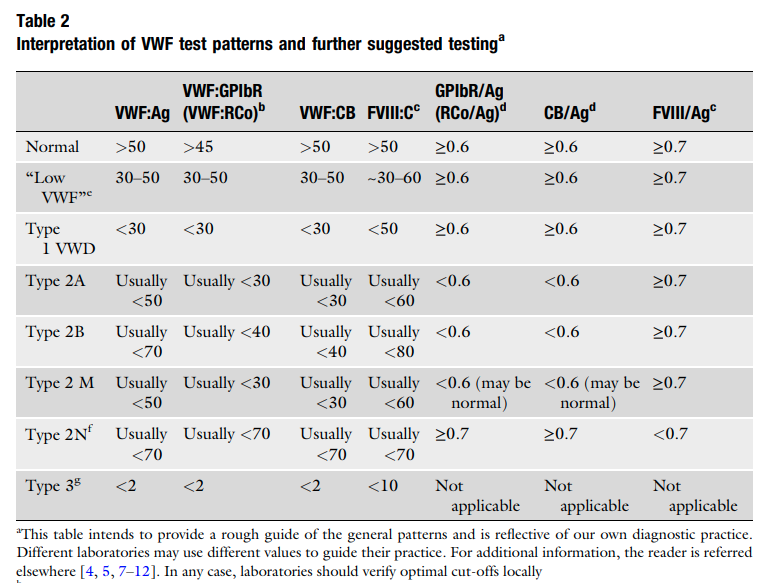
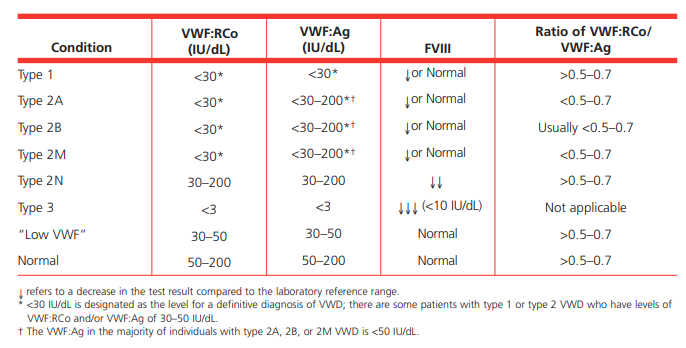
- Suspect diagnosis of type 2 VWD in a case of autosomal recessive inheritance with disproportionately low FVIII:C levels compared with VWF levels (FVIII:C/VWF:Ag ratio< 0.5).1617
- Confirm diagnosis with either:18
- Measurement of the capacity of VWF to bind FVIII (VWF:FVIIIB):
- VWF:FVIIIB is an ELISA method that measures in patient plasma the binding affinity to VWF of added recombinant FVIII (rFVIII).
- Performance of the VWF:FVIII binding assay helps differentiate type 2N VWD and hemophilia A.
- Homozygotes or compound heterozygotes for type 2N exhibit no or severely reduced VWF:FVIIIB (<15%) and very low ratios of VWF:FVIIIB/VWF:Ag (<0.3).
- In heterozygotes VWF:FVIIIB is moderately reduced or normal but the VWF:FVIIIB/VWF:Ag ratios are below the normal range ( <75%)
- Performed only in a relatively small number of specialized laboratories
- DNA sequencing:
- Measurement of the capacity of VWF to bind FVIII (VWF:FVIIIB):
- Other VWF-related measurements are typically normal including:19
- VWF:Ag
- Ristocetin cofactor activity (VWF:RCo)
- Collagen binding activity (VWF:CB)
- Multimeric pattern


Treatment
- DDAVP:20
- A synthetic analogue of the antidiuretic hormone vasopressin.
- Causes release of endogenous VWF and FVIII from vascular endothelial cells into the circulation.
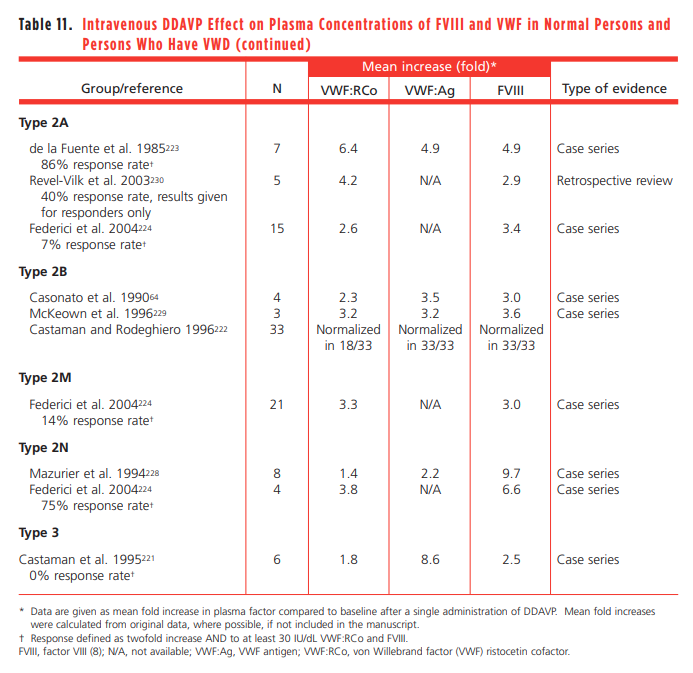
- In type 2N associated with homozygous or heterozygous R854Q mutation (by far the most frequent mutation), desmopressin is usually able to correct FVIII deficiency, although its half-life may be relatively short.21
- In patients with type 2N VWD, DDAVP usually produces an increase in plasma FVIII:C, but fails to correct the capacity of VWF to bind endogenous FVIII; unable to maintain for an adequate time period hemostatic FVIII:C levels owing to the short plasma half-life of this moiety.
- Individuals who have type 2N VWD exhibit a brisk rise in plasma FVIII after receiving DDAVP, but they have a mean FVIII half-life of only 3 hours because of deficient FVIII stabilization by the defective VWF.
- Explorative trial is recommended, with blood samples taken at baseline, 1, and 4 h following administration in order to evaluate not only the peak but also and most importantly the clearance pattern of FVIII:C.22
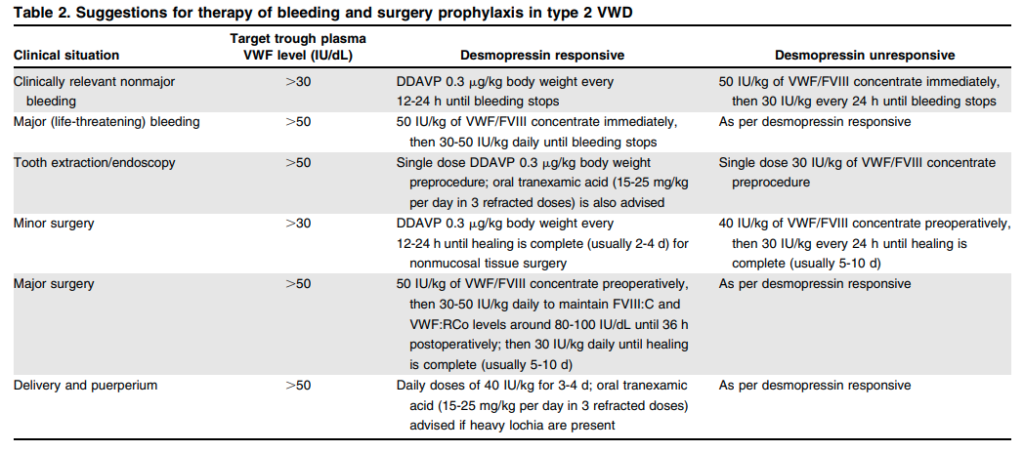
- Replacement therapy:
- Concentrates that contain both VWF and FVIII, including:23
- Humate-P:24
- A lyophilized concentrate of purified VWF and FVIII
- Contains other plasma proteins including fibrinogen and albumin
- When reconstituted at the recommended volume, each milliliter of the product contains 50–100 IU/mL VWF:RCo and 20–40 IU/mL FVIII activity.
- The ratio of VWF:RCo to FVIII for Humate-P in various reports is 2.7, 2, and 1.6.
- Alphanate
- A lyophilized concentrate of VWF and FVIII, and other plasma proteins.
- Prepared from pooled human plasma by cryoprecipitation of FVIII, fractional solubilization, and further purification employing heparin-coupled, cross-linked agarose.
- Upon reconstitution to the recommended volume, each milliliter of product contains 40-180 IU/mL FVIII activity, and not less than 16 IU/mL VWF:RCo activity.
- Wilate
- Humate-P:24
- Administration of FVIII alone will result in its rapid degradation as long as there is no functional VWF.
- Administration of VWF alone may lead to early treatment failure as the liver synthesizes new FVIII.
- Expert opinion:
- Up-to-Date:
- Treatment of type 2N VWD requires administration of VWF concentrates (which bind factor VIII normally), restoring the normal plasma level and half-life of FVIII. The endogenous production of factor VIII is normal in these patients.
- These individuals with low FVIII initially need to receive concentrates of both factor VIII and VWF for bleeding or major surgery. Since they are able to produce endogenous factor VIII normally, subsequent doses of VWF do not need to contain factor VIII, and either plasma-derived VWF concentrates or recombinant VWF concentrates may be used. FVIII levels should be followed and maintained at 50 to 100 IU/dL for the duration of therapy.
- Recombinant FVIII has been used for surgery; individuals with type 2N VWD require FVIII with the first rVWF dose.
- Up-to-Date:
- Concentrates that contain both VWF and FVIII, including:23
- Pregnancy:25
- During pregnancy there is an increase in both VWF and FVIII levels, but in patients with type 2 VWD, FVIII levels may remain low owing to the impaired binding of this moiety by the still dysfunctional albeit increased VWF.
- Patients with VWD type 2N associated with the common R854Q mutation often show a complete normalization of FVIII:C, and no treatment is usually required.26
- Expert opinion:
- Seidizadeh et al:
- During pregnancy it is recommended to use VWF-containing products in patients with FVIII levels lower than 40 IU/ dl.
- Postpartum factor replacement is recommended for at least 3 to 4 days after vaginal delivery and 5 to 7 days after Cesarean section.
- Castaman and James, 2019:
- In pregnant women with type 2N VWD:
- Normalization of FVIII, which is more reduced compared to VWF in this type, usually occurs during pregnancy in women heterozygous or homozygous for R854Q, similar to what happens after desmopressin.
- Usually, these women can be safely managed with desmopressin in case of bleeding complications.
- The results of a desmopressin trial before pregnancy are helpful to predict its usefulness at parturition.
- Information about the more rare mutations is lacking and patients compound heterozygous with a null allele usually do not achieve a normal FVIII level and thus need treatment with VWF concentrates to stabilize FVIII level for a longer period:
- During labor and before epidural anesthesia, 50 IU/kg of VWF should be administered, followed by 30–40 IU/kg/daily for at least 3 days.
- Daily monitoring of FVIII and VWF activity is recommended during the same period.
- Oral tranexamic acid is advised (15–25 mg/kg) up to 10–15 days.
- In pregnant women with type 2N VWD:
- Tosetto and Castaman, 2015:
- Pregnant VWD patients should be monitored for VWF:RCo and FVIII:C at least once during the third trimester of pregnancy.
- No intervention is recommended when FVIII:C and VWF:RCo levels are both around or higher than 50 IU/dL
- If responsive to DDAVP, women with levels only slightly above or around 50 IU/dL may receive a single dose of desmopressin at the beginning of labor, especially if epidural anesthesia is required.
- Seidizadeh et al:

Guideline recommendations27
- ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease

- British Journal of Haematology 2021 guidelines on the clinical and laboratory diagnosis of heritable platelet disorders in adults and children::
