Note: until the discovery and incorporation of ADAMTS13 into the definition of TTP, it is not possible to know how many of the previous cases in the literature were really TTP vs. another type of thrombotic microangiopathy (TMA).
Introduction
The history of thrombotic thrombocytopenic purpura (TTP) reflects the evolving interplay between careful bedside observation and advances in laboratory science. First described in 1924 by Dr. Eli Moschcowitz, who reported a young woman with fever, anemia, petechiae, neurologic changes, and renal failure, the syndrome was initially almost uniformly fatal. Over the decades, case reports and autopsy findings deepened understanding of the microvascular thrombi that define the disease. In the 1970s, the introduction of plasma exchange transformed TTP from a nearly always fatal disorder into a largely treatable one. By the late 1990s, the discovery that severe deficiency of the metalloprotease ADAMTS13 underlies most cases of immune-mediated TTP provided a unifying pathophysiologic explanation and opened the door to targeted therapies. The trajectory of TTP—from mysterious and lethal illness to a mechanistically understood, treatable condition—captures the broader arc of modern medicine: observation, intervention, discovery, and ongoing refinement.
In this section, we are not taking a narrative or highly editorial approach to history. Instead, we frame the story through the raw data that evolved over time—a review of the landmark studies that shaped our current understanding of TTP. Some passages are presented verbatim from the original reports, both to preserve the authenticity of these descriptions and to avoid the risk of misrepresentation that might come with paraphrasing. This risk is heightened by the fact that earlier authors wrote in different eras, with distinct styles, grammar, and medical lexicon that do not always translate neatly into modern terms.
It is also important to retain the original wording because any consideration of the history of medicine must take into account the investigators’ thought processes and how their findings and conclusions were iteratively built upon by subsequent researchers. In that spirit, many of the introduction and conclusion sections are quoted verbatim, as these are the places where the authors themselves editorialized and where one can most clearly see their intellectual grounding, their goals, and their mission. Readers are encouraged to draw their own lessons from this remarkable journey into the past.
1925 – Index Case Published

- Moschcowitz, 1925:1
- Initial description of a patient with TTP (index case):
- 16-year-old female became acutely ill from otherwise previously good health.
- Complained of:
- Weakness in the upper extremities
- Pain on moving the wrists and elbows
- Constant fever up to 104 F
- Physical exam revealed a few petechiae on the left arm.
- The red blood count was 1,330,000/ul; the hemoglobin 40% [thus, anemic].
- Platelet count was not performed.
- While in hospital she developed partial paresis of the left arm and leg; also, a slight facial paralysis.
- She went into a coma and died after a short hospital stay (day 4).
- Postmortem examination demonstrated widespread microvascular thrombi involving the heart, kidneys, and the spleen (“With the low power of the microscope, practically every field revealed from one to a dozen structures that were unquestionably thrombi in the terminal arterioles or capillaries”).
- Moschcowitz concluded:
- “A fairly complete search of the medical literature fails to reveal a case resembling this, either clinically or anatomically. Hyaline thrombi have long been recognized but never in respect to the enormous spread and distribution revealed in this case”.
- “I have learned that Dr. Max Lederer of Brooklyn has seen four cases [apparently never published] clinically identical with the one described in this report. He permits me to state that, thus far, no cause has been found, and that all four patients recovered promptly after a single transfusion of blood”.
- “From these observations we conclude that death, in the case described, resulted from some powerful poison which had both agglutinative and hemolytic properties. If opportunity offers, further investigation will be made of this strange disease with strange pathologic morphology”.
- Initial description of a patient with TTP (index case):

Cataland SR et al. Blood. 2024 Sep 12;144(11):1143-1152
1947 – TTP is Named

- Singer et al, 1947:
- Reported on a case of an 11-year-old female:
- Presented with an upper respiratory infection with a low-grade fever.
- “She seemed to recover after 3 days, but one week later she was extremely tired and listless“.
- On physical exam, she had a fever (100-102.2 F) and petechiae and bruises on the extremities.
- Labs revealed:
- Hb 7 g/dL (anemia)
- Platelets 21,000 (thrombocytopenia)
- Blood smear with poikilocytosis [presumably schistocytes] and polychromatophilia
- “The child’s course was progressively downhill”.
- “On the third day, the patient suddenly became incoherent, complained of dizziness and had paresthesias in all extremities. On the sixth day after admission, the patient lapsed into coma and died“.
- Autopsy: Heart showed innumerable thrombi were seen throughout the sections. They were located in the capillaries, arterioles, and in smaller arteries. The thrombi consisted mainly of thrombocytes and some fibrin. Platelet thrombi were also noted in the spleen, kidneys, urinary bladder, suprarenals, pancreas, and brain.
- Conclusions:
- “In comparing the clinical and histologic features of our case with those of the 11 previously reported ones almost identical clinical and pathologic findings are encountered. From this it is quite obvious that we are dealing with a definite disease entity“.
- “In order to make a clinical diagnosis of this disease it is imperative that the diagnostician consider this rare disorder when confronted with a syndrome of thrombocytopenic purpura. A glance at the list of references will demonstrate that no distinctive name has been attached to this disorder until now. The term ‘generalized arteriolar and capillary platelet thrombosis’ is too long and cumbersome for practical use. We suggest the name “thrombotic thrombocytopenic purpura” for this syndrome, using the word ‘thrombotic’ in the sense of ’caused by thrombosis'”.
- Thrombocytopenia resulted from a “disappearance of the platelets from the circulation due to the formation of the myriads of platelet thrombi”.
- Reported on a case of an 11-year-old female:

1959 – Exchange Transfusion to Treat TTP

- Rubinstein et al, 1959:
- Introduction (verbatim):
- Refers to 78 cases of TTP reported in the available literature.
- The anemia has been shown to be hemolytic, and the hemorrhagic tendency to be due to thrombocytopenia. However, the nature of the characteristic vascular thrombi has not been definitely elucidated. First attributed to erythrocytes, then thought to be composed of agglutinated platelets, the thrombi are now regarded largely as the result of endothelial lesions, conceived as the primary “prethrombotic” factor, with possible secondary platelet deposition”.
- According to certain investigators, the vascular changes leading to formation of minute arteriolar and capillary aneurysms constitute the fundamental lesion.
- The syndrome is characterized by:
- Marked hemolytic anemia
- Thrombocytopenia
- Signs of cerebral involvement which are at first transient
- The disease usually has a sudden onset, with fever, prostration, the appearance of jaundice (usually mild), and purpura, mental confusion, and changing neurologic signs. Most patients have a very short, fatal course; rarely, there may be a more protracted course, and the full-blown picture may develop only after some time.
- While the etiology remains unknown, it “appears to be caused by a common immunologic mechanism“.
- No effective treatment is available.
- Case:
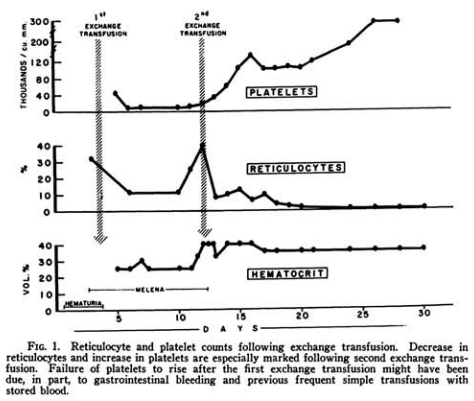
- “We have observed a case that fits the clinical criteria of TTP mentioned above, and that is reported because of the unusual clinical response following exchange transfusions using fresh whole blood“.
- An 11-year-old white girl presented with sudden onset of fever, nausea and vomiting, and difficulty in speech.
- The next day there was some mental confusion.
- Physical exam revealed mild jaundice and small petechiae over the legs and forearms.
- Labs showed:
- Hemoglobin 4.7 gm.% (anemia)
- White blood cells 14,200
- Blood smear with about 30% polychromatophilic cells, and “about the same number to be small spherocytes, giving a biphasic character to the red cell population. In addition, the red cells showed numerous instances of fragmentation. The reticulocyte count was 34%. These findings indicated increased hemolysis“.
- The platelet count was between 80,000 and 20,000 per cubic millimeter (thrombocytopenia).
- The patient was treated with steroids.
- In addition, she received to blood exchange transfusions:
- The first with 2,000 c.c. of fresh whole blood
- The second with 3,000 c.c. of whole blood clinical
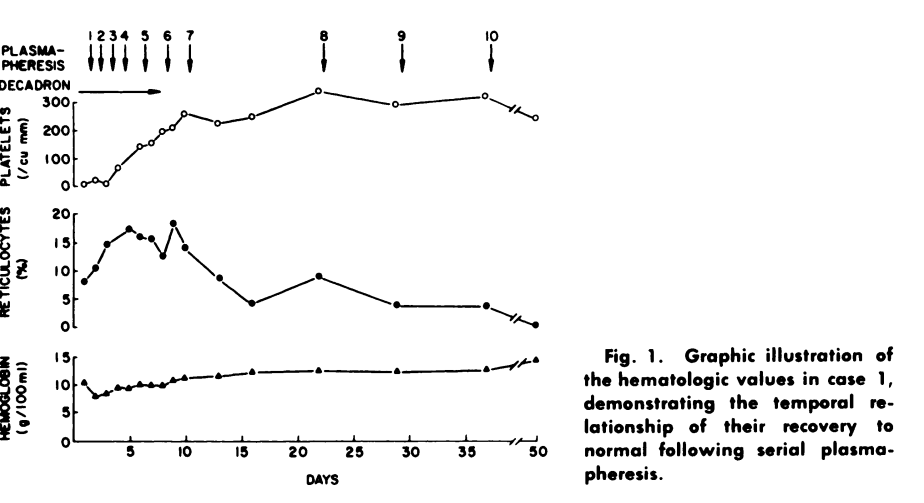
- She demonstrated some hematologic improvement (see figure below).
- Conclusions (verbatim):
- The mechanism of the benefits of the exchange transfusions is not clear. One may speculate that circulating antibodies were removed, while formation of new antibodies was inhibited by the previously administered high dosages of cortisone and ACTH.
- This case is reported because of the unusual therapeutic response to exchange transfusions with fresh blood, a hitherto unreported procedure in thrombotic thrombocytopenic purpura.
- Introduction (verbatim):

1966 – Review of 271 Cases of TTP
Note: All of these cases occurred before the discovery of ADAMTS13. It is difficult to know how many of them would have been defined as TTP by today’s standards!

- Amorosi and Ultmann, 1966:2
- Introduction:
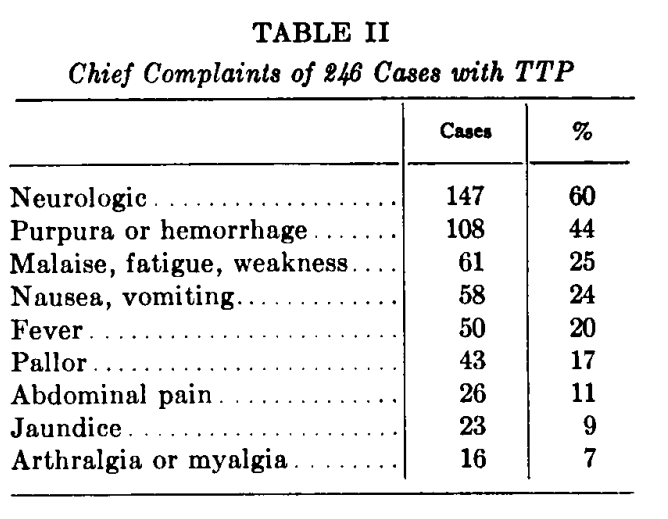
- Authors state that 271 cases of TTP had been reported in the literature since the original description in 1925.
- Authors refer to names used to describe the condition:
- Moschcowitz’s disease
- TTP
- Thrombotic microangiopathic hemolytic anemia
- Thrombocytic acro-angiothrombosis
- Disseminated arteriolar and capillary thromboses
- Platelet thrombosis syndrome
- Thrombohemolytic thrombocytopenic purpura
- TTP is usually described as a triad of clinical findings:
- Thrombocytopenic purpura
- Hemolytic anemia
- Neurological manifestations
- However, fever and renal disease “almost invariably present”. Thus, “the syndrome should be considered as a pentad of findings.
- Diagnostic confirmation requires histologic demonstration of widespread hyaline occlusion of terminal arterioles and capillaries.
- The paper provides detailed analysis of 16 of the authors’ own patients (13 of whom had been previously reported) and a review of cases from the literature.
- Results:
- Findings are shown from the original paper:
- Introduction:
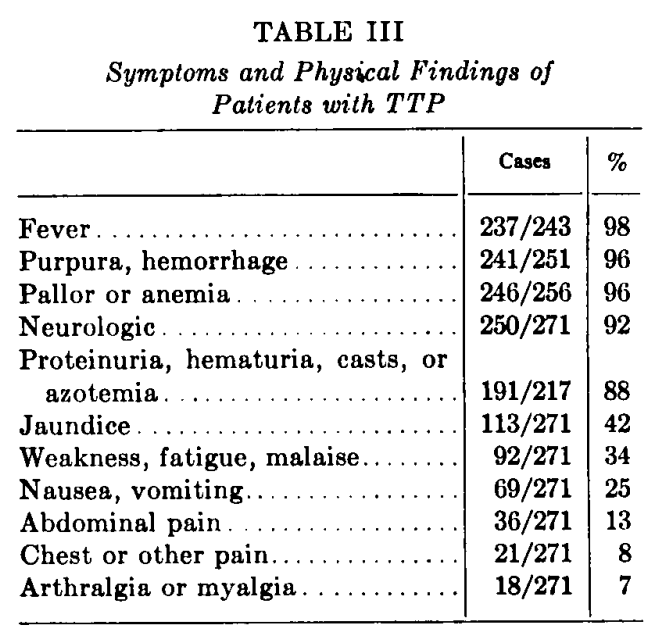
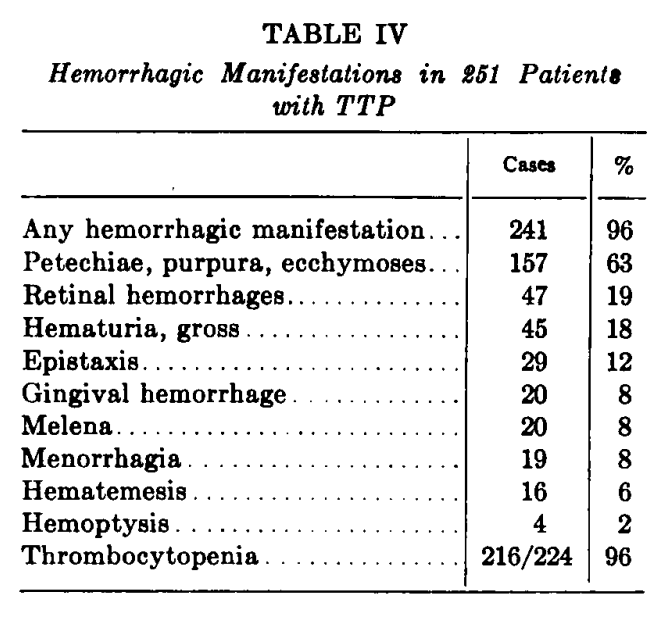



- Amorosi and Ultmann, 1966 (cont’d):
- Results (cont’d):
- Postmortem:
- Characteristic vascular occlusions may be found in many organs, but most commonly seen in:
- Heart
- Brain
- Kidneys
- Pancreas
- Adrenals
- Striking absence of inflammatory change in the involved vessels.
- Affects primarily capillaries and arterioles.
- Associated with amorphous hyaline-like material accompanied by endothelial proliferation.
- “The nature of the occluding material is still in doubt”. Originally thought to represent agglutinated red cells or platelets.
- Characteristic vascular occlusions may be found in many organs, but most commonly seen in:
- Postmortem:
- Conclusions about etiology:
- “A number of theories have been proposed”.
- “It’s etiology or etiologies, however, remain unknown”.
- Among the causes that have been implicated include:
- Toxins
- Drug sensitivity
- Bacterial infections
- Autoimmune reactions
- Collagen disease
- Abnormalities in serum lipids
- Results (cont’d):

1977 – First use of Plasmapheresis in TTP

- Bukowski et al, 1977:
- Introduction:
- The therapeutic regimens employed to date have included:
- High-dose corticosteroids
- Splenectomy
- Heparin
- Antiplatelet agents
- Dextran
- Urokinase
- Whole blood exchange transfusions
- “During the past 16 yr, we have treated 15 patients with exchange transfusions,3 and have noted complete and lasting remissions in 9 instances”.
- “The possibility that these remissions may be related to removal of soluble toxic material has led to the use of plasmapheresis in this disorder”.
- Cases:
- Two cases of TTP were reported.
- One was treated with corticosteroids.
- Both received serial plasmapheresis (each patient received a total of 10).
- Conclusions (verbatim):
- The mode of action of exchange transfusions is unknown but may be related to removal of a circulating toxic substance.
- Recent speculation that the pathogenesis of some cases of TTP may involve soluble immune complexes and our experience with exchange transfusions has led to our use of plasmapheresis in this syndrome.
- Plasmapheresis has been reported to remove circulating antibody or immune complexes in a wide variety of immunologic disorders such as Goodpasture syndrome and systemic lupus erythematosus. Whether the mode of action of exchange transfusions and plasmapheresis in patients with TTP is analogous to this is unknown.
- The exact value of plasmapheresis compared to exchange transfusions in this syndrome is unknown. Plasmapheresis appears to be a simpler method of therapy and does not require large amounts of fresh whole blood as do exchange transfusion.
- The therapeutic regimens employed to date have included:
- Introduction:


1982 – Unusually Large vWF multimers in TTP

- Moake et al, 1982
- Introduction:
- A platelet agglutinating factor has been detected in the plasma of some patients with TTP.
- The characteristics of the agglutination activity reminded the authors of those of large multimeric vWF components and led to this study to investigate the properties of plasma vWF in patients with TTP.
- They studied the plasma from 4 patients with chronic relapsing forms of TTP.
- Results:
- vWF multimers larger than those present in normal plasma were found in plasma from the four patients.
- They were most apparent during remission.
- Relapses were associated with decreased multimers, which may “indicate consumption of the large [vWF multimers] during relapse”.
- Conclusions (verbatim):
- The fundamental defect associated with susceptibility to recurrent episodes of TTP remains to be identified precisely. It may be congenital or possibly acquired. The missing activity may be a large VIII:vWF depolymerase … reduces the size of the very large VIII:vWF multimers synthesized and secreted into the circulating blood by the endothelial cells.
- During severe episodes of TTP, partial removal of the unusually large VIII:vWF multimers (and possible the inciting cofactor) by plasmapheresis, as well as transfusion of normal plasma, may be required to control in vitro platelet agglutination.
- Introduction:

1991 – Steroids vs. Plasmapheresis in TTP

- Bell et al, 1991
- Introduction:
- TTP and HUS are regarded as parts of a spectrum often designated TTP-HUS.
- Initial papers reported mortality rate of 100%.
- One of the first documented survivors was treated with exchange transfusions.
- “In 1963, it was suggested that plasma therapy may be beneficial”.4
- “We report our experience with the treatment of TTP-HUS with corticosteroids and plasmapheresis with plasma replacement”.
- “A placebo-controlled study was not done because plasmapheresis is the only form of therapy for TTP-HUS that is acknowledged to be effective”.
- Methods:
- Retrospective study
- Diagnosis based on patients having at least 4 of the following:
- MAHA with negative Coombs test
- Thrombocytopenia
- CNS abnormalities
- Fever
- Renal dysfunction
- Treatment:
- If patient had minimal symptoms and was free of CNS findings, corticosteroids alone (prednisone 200 mg/day); switched to plasmapheresis if CNS findings, no clinical improvement or deterioration within 48 h. Steroids were continued.
- If patient had moderate to severe symptoms with CNS changes or rapid decline, they received plasmapheresis once daily
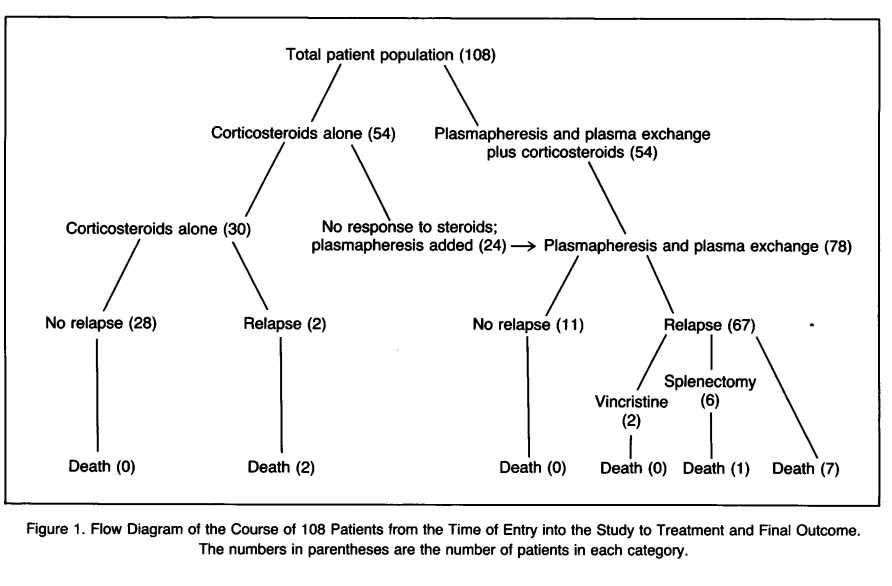
- Results:
- Total of 108 patients
- Prednisone alone effective in 30 patients with mild TTP-HUS, 2 relapses and 2 deaths
- Prednisone + TPE given in 78 patients with complicated TTP-HUS – 67 relapses and 8 deaths
- 10 deaths (9% mortality)
- Conclusions (verbatim):
- “The observations that some form of plasma therapy is beneficial seems valid”.
- “Some investigators subsequently suggested that infusion of FFP was as efficacious as plasmapheresis and plasma exchange. It is not apparent that the infusion of FFP alone is not efficacious for the majority of patients“.
- “Explanation for its therapeutic success [of plasmapheresis] remains unknown”.
- “Our results suggest that it is reasonable to employ corticosteroids in the initial treatment of this illness”.
- Introduction:
1991 – Plasma Exchange vs. Plasmapheresis in TTP

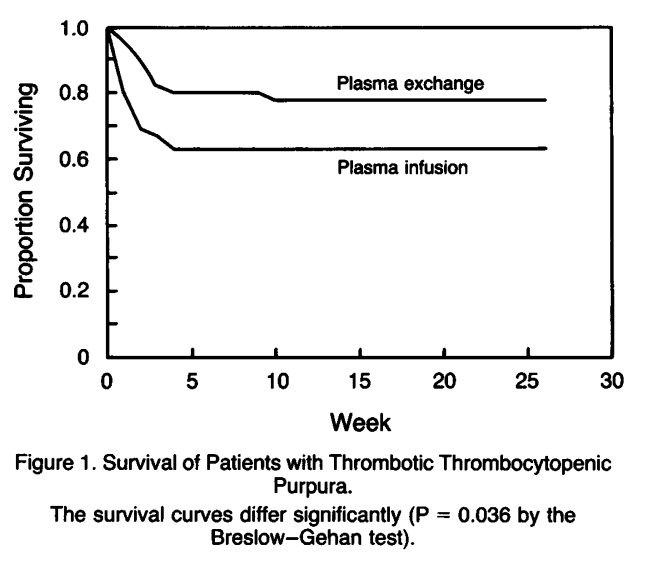
- Rock et al, 1991:
- 102 patients with thrombotic thrombocytopenic purpura
- Randomly assigned to receive either:
- Plasma exchange
- Plasma infusion
- All patients also received aspirin and dipyridamole.
- The outcomes in the two groups were compared at the end of the first treatment cycle (day 9) and after six months.
- At the end of the first treatment cycle:
- Compared to plasma infusions, those receiving plasma exchange had:
- Higher rate of response as defined by an increase in the platelet count (P = 0.025).
- Lower mortality (P = 0.035).
- Compared to plasma infusions, those receiving plasma exchange had:
- After six months:
- Compared to plasma infusions, those receiving plasma exchange had:
- Higher rate of response as defined by an increase in the platelet count (P = 0.002).
- Lower mortality (P = 0.036).
- The overall mortality was 29 percent.
- Compared to plasma infusions, those receiving plasma exchange had:
- Conclusions: Plasma exchange is more effective than plasma infusion in the treatment of thrombotic thrombocytopenic purpura.

For Journal Club of this study, click here.
1996 – Discovery of vWF-Cleaving Protease
The initial term for ADAMTS13

- Furlan et al, 1996
- Introduction (verbatim):
- Unusually large molecular forms of vWF were found in patients with TTP.5
- These abnormally large multimers disappeared after transfusion of normal fresh frozen plasma.
- It is evident that proteolytic enzyme(s) may be involved in the physiological regulation of the polymeric size of vWF in the circulating blood and may also play an important role in the pathogenesis of vWF abnormalities in some patients with congenital and acquired disorders of hemostasis.
- The proteolytic enzyme responsible for vWF degradation in normal human plasma has not yet been identified.
- In the present report, we attempted to purify from normal human plasma a protease degrading vWF to LMW [low molecular weight] forms and producing peptide fragments physiologically occurring in circulating blood. The results of our studies provide evidence that the proteolytic activity, cleaving the peptide bond 842Tyr-843Met in the vWF subunit, is associated with a HMW protein that is different from known serine proteases, cathepsins, matrix metalloproteinases, and calpains.
- Results:
- A vWF-degrading protease was purified (10,000-fold) from human plasma using chelating Sepharose, hydrophobic interaction chromatography, and gel filtration.
- The enzyme was found to be virtually absent in the platelet lysates obtained by repeated freezing and thawing.
- The proteolytic activity was associated with a high molecular weight protein (-300 kD) as judged by gel filtration and sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
- vWF was resistant against the protease in a neutral buffer at physiological ionic strength but became degraded at low salt concentration or in the presence of 1 mol/L urea.
- No degradation of human fibrinogen, bovine serum albumin, or calf skin collagen by the purified protease was noted under the same experimental conditions.
- The observed properties of the vWF-degrading enzyme differ from those of all other previously described proteases.
- “Purified vWF was incubated with the protease, and the degraded material subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis after disulfide reduction. The size, amino acid composition, and amino terminal sequence of the reduced fragments confirmed that the peptide bond 842Tyr-843Met had been cleaved, i.e., the same bond that has been proposed to be cleaved in vivo”.
- Conclusions:
- “In conclusion, our results show that the partially purified protease cleaves the peptide bond 842Tyr-843Met [of vWF] that is assumed to be split in vivo. It is conceivable that the protease described in this communication may be involved in the regulation of the polymeric size of vWF in the circulating blood and, thus, may affect primary hemostasis”.
- Introduction (verbatim):

- Tsai, HM, 1996
- Introduction (verbatim):
- As a secretory protein, vWF is distinctive in that it exists in the plasma as a series of multimers with molecular weights ranging up to approximately 12,000 kD. Several lines of evidence suggest that molecular size is a major determinant of vWF capability in supporting platelet adhesion and aggregation. Although vWF multimers were originally believed to be secreted from endothelial cells, recent studies show that vWF is secreted from endothelial cells as an extra-large polymer.
- vWF is subjected in the circulation to proteolysis.
- In the course of investigating the mechanisms involved in proteolytic cleavage of vWF, we reported that shear stress on vWF promoted its cleavage by a EGTA-inhibited protease in normal plasma. In this study, we partially purified and characterized the protease and further investigated the role of vWF conformation in its susceptibility to the protease.
- Results:
- he plasma protease was enriched nearly 900-fold using a sequence of purification steps, including Sephacryl S-300 HR gel filtration, Matrex gel orange A dye affinity chromatography, and Q Sepharose anion exchange chromatography. The protein had an estimated molecular mass of ~200 kDa and was inhibited by EDTA, EGTA, and 1,10-phenanthroline.
- Chelation with EGTA or EDTA blocked the enzyme’s activity, but this effect could be reversed with calcium (Ca²⁺), though not with magnesium (Mg²⁺).
- The protease was not affected by a wide range of synthetic or natural protease inhibitors, and it did not bind to gelatin-agarose.
- Activity was detected even in plasma lacking various coagulation or anticoagulation proteins, indicating independence from those pathways.
- When von Willebrand factor (vWF) was cleaved by the protease, the resulting fragments (identified by peptide-specific antibodies VP-1 and LJ-7745) matched those occurring naturally in plasma, and were distinct from fragments generated by plasmin.
- Large endothelial vWF multimers, once exposed to guanidine HCl or high shear stress, were susceptible to cleavage by the protease into smaller fragments.
- Conclusions:
- These results support the model that endothelial secreted vWF is converted to multimers by a novel plasma metalloproteinase.
- Although native vWF exists in a conformation relatively resistant to cleavage, an alteration in the conformation by shear stress can lead to an enhanced proteolytic susceptibility.
- This model may explain the decrease in vWF multimer sizes in various clinical conditions.
- Introduction (verbatim):

1997 – Deficient activity of von Willebrand factor‐cleaving protease in TTP

- Furlan et al, 1997
- Introduction:
- In patients with TTP, excessive intravascular platelet aggregation has been associated with appearance in plasma of unusually large vWF multimers.
- These extremely adhesive vWF multimers may arise due to deficiency of a “depolymerase” cleaving vWF to smaller molecular forms, either by reducing the interdimeric disulfide bridges or by proteolytic degradation.
- We studied the activity of a recently described vWF-cleaving protease in four patients with chronic relapsing TTP.
- Results:
- Diluted plasma samples of TTP patients were incubated with purified normal human vWF in the presence of a serine protease inhibitor, at low ionic strength, and in the presence of urea and barium ions.
- The extent of vWF degradation was assayed by electrophoresis in sodium dodecyl sulfate-agarose gels and immunoblotting.
- Four patients, that included two brothers, with chronic relapsing TTP displayed either substantially reduced levels or a complete absence of vWF-cleaving protease activity. In none of these patient plasmas was an inhibitor of or an antibody against the vWF-cleaving protease established.
- Conclusions:
- “Our data suggest that the unusually large vWF multimers found in TTP patients may be caused by deficient vWF-cleaving protease activity. Deficiency of this protease may be inherited in an autosomal recessive manner and seems to predispose to chronic relapsing TTP. The assay of the vWF-cleaving protease activity may be used as a sensitive diagnostic tool for identification of subjects with a latent TTP tendency”.
- Introduction:
2001 – Partial Amino Acid Sequence of vWF-CP

- Gerritsen et al, 2001
- Introduction:
- von Willebrand factor-cleaving protease (vWF-cp) is responsible for the continuous degradation of plasma vWF multimers released from endothelial cells.
- vWF-cp is deficient in patients with thrombotic thrombocytopenic purpura, who show unusually large vWF multimers in plasma.
- Purified vWF-cp may be useful for replacement in these patients, who are now treated by plasma therapy.
- In this study, vWF-cp was purified from normal human plasma by affinity chromatography of the IgG fraction from a patient with autoantibodies to vWF-cp and by a series of further chromatographic procedures, including affinity chromatography on Protein G, Ig-TheraSorb, lentil lectin, and heparin.
- Results:
- Under nonreducing SDS-PAGE conditions, single-chain protein bands of approximately 150, 140, 130, and 110 kDa were observed. Each displayed the same N-terminal amino acid sequence, indicating they originated from a common polypeptide that had undergone partial degradation at the C-terminal end.
- The N-terminal sequence of the first 15 amino acids was identified as: Ala–Ala–Gly–Gly–Ile–Leu–His–Leu–Glu–Leu–Leu–Val–Ala–Val–Gly.
- In gel filtration studies, the protease was found to migrate as part of a high-molecular-weight complex with clusterin, a ~70 kDa protein with chaperone-like properties. This association could be disrupted by concentrated chaotropic salts.
- In contrast, in normal human plasma or serum, vWF-cleaving protease (vWF-cp) was not bound to clusterin, suggesting that the complex seen during purification was an artifact caused by partial denaturation of vWF-cp.
- The activity of vWF-cp was remarkably stable at 37 °C.
- In vitro, the protease maintained activity for more than a week in citrated plasma, heparinized plasma, or serum.
- A transient increase in activity was noted during the first three days of incubation.
- Conclusions (verbatim):
- Unusually large vWF multimers were detected in patients with TTP, and increased vWF binding to platelets was demonstrated in TTP patients.
- In 1996, 2 groups of investigators independently identified and partially purified a protease from normal human plasma that was shown to be responsible for in vivo degradation of vWF multimers.
- Severe congenital deficiency of this protease was established in patients with familial TTP, and the presence of vWF-cp–inhibiting autoantibodies was observed in patients with nonfamilial TTP.
- VWF-cp consists of 1427 amino acid residues and has a signal peptide, a short propeptide terminating in the sequence RQRR, a reprolysin-like metalloprotease domain, a disintegrin-like domain, a thrombospondin-1 repeat, a Cys-rich domain, an ADAMTS spacer, seven additional thrombospondin-1 repeats, and two CUB domains.
- Note added in proof: “Note added in proof. “The reader is referred to the accompanying paper, Fujikawa et al, characterizing vWF-cp as a metalloprotease.”
- Introduction:

- Fujikawa et al, 2001
- Introduction:
- vWF is synthesized in megakaryocytes and endothelial cells as a very large multimer, but it circulates in plasma as a group of multimers ranging from 500 to 10 000 Kd.
- An important mechanism for depolymerization of the large multimers is the limited proteolysis by a vWF-cleaving protease present in plasma.
- The absence or inactivation of the vWF-cleaving protease results in the accumulation of large multimers, which may cause thrombotic thrombocytopenic purpura.
- Furlan et al, and Tsai [see studies above) reported the partial purification of the vWF-cleaving protease from human serum in 1996:
- The apparent molecular weight of the enzyme was approximately 300 Kd.
- The enzyme required divalent cations for catalytic activity.
- The enzyme was found to specifically cleave the Y(842)–M(843) bond to produce the 140- and 170-kd bands, which were the same as those present in plasma-derived vWF.
- Results:
- SDS-PAGE analysis of fractions from anion exchange and gel filtration chromatography demonstrated that the vWF-cleaving protease activity was associated with a protein band of approximately 150 kDa.
- Under reducing conditions, the same protein migrated with an apparent molecular weight of about 190 kDa.
- Sequencing of the amino terminus of the 150-kDa band yielded the sequence AAGGIL(H)LE(L)L(D)AXG(P)X(V)XQ, with several positions only tentatively identified.
- A search of the human genome sequence identified the vWF-cleaving protease as a new member of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin type I motif) family of metalloproteinase.
- Conclusions:
- These analyses show that the vWF-cleaving protease is a new metalloproteinase that belongs to the ADAMTS subfamily.
- Note:
- “In the accompanying paper (see Gerritsen et al above) the authors also purified vWF-cleaving protease to apparent homogeneity. The amino-terminal sequence was essentially identical to that reported in this paper”.
- Introduction:

2001 – Cloning of ADAMTS13

- Levy et al, 2001
- Introduction (verbatim):
- “Although treatment outcome has improved significantly, the molecular pathogenesis of TTP is still unknown and the specific plasma factor(s) responsible for the acute onset of this disease, or recovery after treatment, remains to be identified”.
- “Unusually large multimeric forms of the platelet-adhesive blood-coagulation protein VWF have been observed in the plasma of TTP patients and are proposed to have a pathogenic role in the formation of the microvascular VWF- and platelet-rich thrombi characteristic of this disorder”.
- “Consistent with this hypothesis, a proteolytic activity that degrades large VWF multimers to smaller sizes has been shown to be decreased in the plasma of TTP patients,6 either on the basis of a constitutional deficiency in congenital cases or the presence of an autoantibody inhibitor in most sporadic adult-onset cases”.
- “These findings suggest that TTP may be triggered by accumulation of large, highly adhesive VWF multimers in the absence of physiologic processing by this VWF-cleaving protease”.
- “However, other studies have implicated platelet-aggregating proteins or endothelial injury as the underlying mechanism and enhanced”.
- Although the VWF-cleaving protease has been partially purified, it seems to be present at relatively low levels in plasma and its identification at the sequence level has remained elusive.
- Results:
- A genetic strategy was applied to study four families with reduced vWF-cleaving protease activity.
- Plasma levels of the protease served as a measurable phenotype for linkage analysis.
- A genome-wide scan was performed using 382 polymorphic microsatellite markers on DNA from affected individuals.
- Using linkage analysis together with cDNA cloning, RT-PCR, RACE, and genomic sequencing, investigators identified a novel zinc metalloproteinase gene of the ADAMTS family (ADAMTS13).
- The full-length ADAMTS13 cDNA sequence (GenBank accession AF414401) and its genomic organization were determined, confirming it encodes a potentially active ADAMTS protease.
- Sequencing of patients’ DNA revealed 12 distinct ADAMTS13 mutations, accounting for 14 of 15 disease alleles studied.
- Conclusions (verbatim):
- We show that deficiency of ADAMTS13 is the molecular mechanism responsible for TTP and suggest that physiologic proteolysis of VWF and/or other ADAMTS13 substrates is required for normal vascular homeostasis.
- Our data provide the first direct evidence of an aetiologic role for VWF-cleaving protease activity in the pathogenesis of TTP and identify the enzyme likely to be responsible for this activity: metalloproteinase ADAMTS13.
- Note added in proof:
- “Fujikawa et al. and Gerritsen et al. have reported purification and sequence analysis of the amino-terminal amino acids of human VWF-cleaving protease. These results confirm the identity of ADAMTS13 as the VWF-cleaving protease and its predicted processing by propeptide convertase cleavage.”
- Introduction (verbatim):

- Zheng et al, 2001
- Introduction:
- The basis for the efficacy of plasma exchange remains unknown, although a plausible model has been suggested in which the proteolysis of VWF plays a central role.
- A plasma metalloprotease was identified that requires both calcium and zinc ions and cleaves the Tyr1605–Met1606 bond2 in the central A2 domain of the VWF subunit.
- Cleavage was stimulated by shear forces like those occurring at sites of arterial thrombosis or by low concentrations of urea or guanidine.
- Furthermore, most adult patients with TTP were found to have congenital deficiency or an acquired autoantibody inhibitor of this VWF-cleaving protease (VWFCP).
- These findings are consistent with the UL-VWF model of TTP pathogenesis.
- VWFCP was recently purified from human plasma sources and an N-terminal amino acid sequence was obtained.
- This information showed it to be a member of the ADAMTS family of metalloproteases, named for the characteristic combination of a disintegrin-like and metalloprotease, with thrombospondin type 1 motif.
- Results:
- The cDNA sequence was determined to be 4.6 kilobase pairs in length.
- Northern blot analysis showed that full-length VWFCP mRNA expression was restricted to the liver.
- The protein product (VWFCP) is composed of 1,427 amino acids and contains multiple domains: a signal peptide, a short propeptide ending in RQRR, a reprolysin-like metalloprotease domain, a disintegrin-like domain, one thrombospondin-1 repeat, a cysteine-rich region, an ADAMTS spacer, seven additional thrombospondin-1 repeats, and two CUB domains.
- The enzyme appears to be synthesized as an inactive precursor (zymogen) that likely undergoes proteolytic activation, possibly mediated by furin within the cell.
- Alternative splicing produces at least seven distinct variants, each truncated at different points beyond the protease domain.
- Conclusions:
- The domain structure of VWFCP indicates that it belongs to the ADAMTS family of metalloproteases, and VWFCP has been designated ADAMTS13.
- Introduction:
Final Thoughts
It is often difficult to know where history ends and present day begins. In this section, we have taken a journey that begins with the first description of TTP in 1947, continues through the transformative discovery that plasma exchange dramatically improves survival, and culminates with the identification of the vWF cleaving protease and subsequent cloning of that protease, later named ADAMTS13, in 2001. Some might argue that 2001 is too recent to count as history, but it is a convenient stopping point because it encapsulates the three most important discoveries in the field: the recognition of the disease, the establishment of its primary treatment, and the identification of the responsible protease that together built the foundation for subsequent therapeutic breakthroughs such as rituximab, caplacizumab, and recombinant ADAMTS13. These advances are amply covered in other tutorials within this TTP module..
Along the way we have seen the discovery of a new disease, the establishment of its mainstay therapy (still in use today), and the unraveling of the underlying enzymatic defect. What is not covered in this section are subsequent therapeutic advances. For those, you can explore the history timeline, which includes: the first case report of rituximab use in 2002, a case series in 2005, a phase 2 study in 2011, and the randomized controlled trial of caplacizumab in 2016. (Caplacizumab is a nanobody directed against the A1 domain of von Willebrand factor that prevents platelet adhesion.) Finally, the story brings us up to the use of recombinant ADAMTS13 in 2024.
I hope you enjoyed this journey through the history of TTP. Now it is time to move on to the other parts of the module.
Want to explore this further?
Check out the related sections in our TTP module:
- PDF of original 1925 article by Eli Moschcowitz
- History of TTP – Timeline Graphic
- Eponyms in Hematology – Upshaw–Schulman syndrome
- Eponyms in Hematology – Moschcowitz syndrome
- Journal Club on Rock et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med. 1991; 325(6): 393-397.
- Journal Club on Scully et al. A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood. 2011;118(7):1746-1753.
- Journal Club on Scully et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019; 380(4): 335-346.
- Journal Club on Scully et al. cTTP Phase 3 Study Investigators. Recombinant ADAMTS13 in Congenital Thrombotic Thrombocytopenic Purpura. N Engl J Med. 2024; 390(17):1584-1596.
{kind=link}
{kind=link}