Overview
- von Willebrand factor (vWF) is a large multimeric adhesive plasma glycoprotein that plays an essential role in normal hemostasis by recruiting platelets to sites of vascular injury (necessary for initial platelet tethering and subsequent platelet adhesion).1
- The function of vWF as a vessel wall damage sensor and initiator of primary hemostasis is highly dependent on its multimeric size. The larger VWF multimers in plasma are the most hemostatically reactive.2
- The activity of vWF is modulated by ADAMTS13, a metalloprotease that cleaves highly procoagulant, ultra-large vWF (UL-VWF) multimers into smaller, less procoagulant forms (i.e., reduces plasma vWF activity). ADAMTS13 (A Disintegrin And Metalloproteinase with ThromboSpondin type 1 repeats, member 13) cleaves vWF at a single peptide bond (Tyr1605-Met1606) located within the central vWF A2 domain, which becomes accessible when vWF unfolds in response to shear stress.
- Thrombotic thrombocytopenic purpura (TTP) is caused by a severe deficiency of the enzyme ADAMTS13, which leads to accumulation of platelet-hyperadhesive UL-VWF multimers on endothelial cells and is clinically manifested by thrombotic microangiopathy (TMA) that is characterized by severe thrombocytopenia, hemolytic anemia, and ischemic organ failure.
von Willebrand factor
- vWF gene and protein:
- The gene encoding vWF:
- Maps to the short arm of chromosome 12 (12p13.31).
- Spans ~176 kb.
- Contains 52 exons.
- vWF cDNA:
- Has a length of ~8.7 kb.
- Encodes a primary translation product (precursor) of 2,813 amino acid residues, which includes:
- 22-amino acid signal peptide
- 741-amino acid VWF propeptide (vWFpp)
- Mature subunit of 2,050 amino acids
- vWF protein:
- Each VWF subunit contains 2,050 amino acid residues with a molecular mass of ~250 kDa under reducing conditions.3
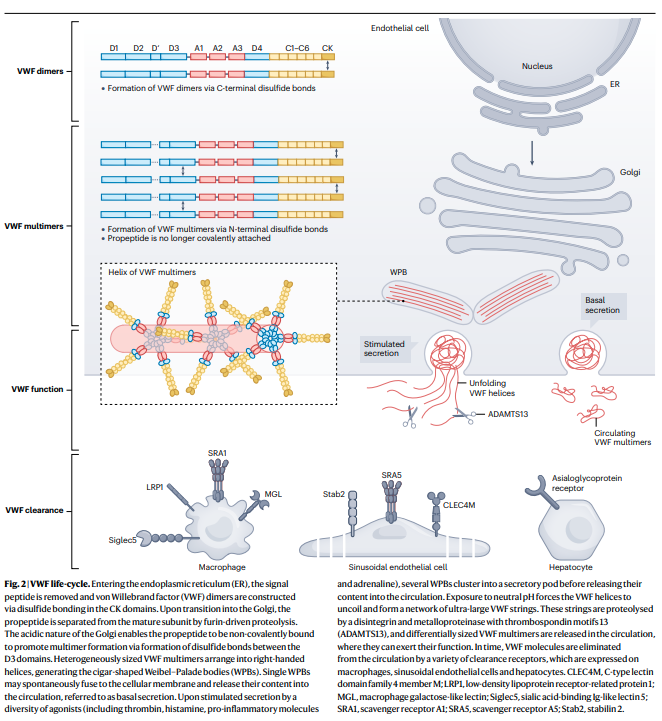
- Composed of structural domains that are repeated or shared with other proteins. These domains are arranged in the following order: D1–D2(VWFpp)–D’–D3–A1–A2–A3–D4–C1–C2–C3–C4–C5–C6–CK.
- The gene encoding vWF:
- Expression of vWF:
- Synthesized in endothelial cells and megakaryocytes.
- Stored in Weibel–Palade bodies (WPBs) in endothelial cells and α-granules in megakaryocytes.
- Multimerization of vWF:
- Initially produced as a single-chain polypeptide with a distinct domain structure, but then undergoes extensive processing to form heterogeneously sized multimeric protein:
- Signal peptide is first removed.
- vWF subunits then dimerize through intermolecular disulfide bridges located in the C-terminal CK domain.
- Finaly, in the Golgi apparatus, vWF multimers form through disulfide bridges between D3 domains:
- First forming nanoclusters of VWF multimers with a median size of 500 nm.
- Next forming mature, larger-sized multimers (high-molecular-weight [ultra-large] VWF multimers).
- Although the largest plasma VWF multimers have generally been considered to range from 20- to 40-mers, more recent measurements have estimated that 100- or 200-mers may also exist.4
- Initially produced as a single-chain polypeptide with a distinct domain structure, but then undergoes extensive processing to form heterogeneously sized multimeric protein:
- Secretion of vWF:
- Basal secretion:
- Single WPBs may spontaneously fuse to the cellular membrane and release their contents into the circulation.
- Basal secretion maintains plasma concentrations of vWF.
- Stimulated secretion:
- vWF release in response to agonists such as:
- Thrombin
- Histamine
- Pro-inflammatory molecules
- Adrenaline
- Several WPBs cluster into a secretory pod before releasing their content into the circulation.
- Leads to immediate release of high local concentrations of ultra-large VWF multimers that can participate in the arrest of bleeding.
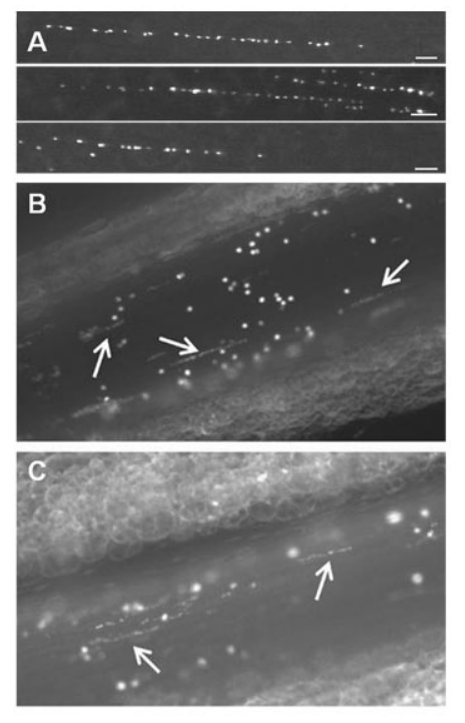
- In contrast to multimers released via basal secretion, stimulated secretion leads to the release of ultra-large vWF multimers that form strings which remain attached to the endothelial cells.5
- As a result of tethering to the endothelium and the rheologic forces of flow, these VWF strings unravel, can bind platelets, and wave in the direction of the blood flow.6
- The strings are susceptible to specific proteolysis by its regulating protease, ADAMTS13. The formation and elongation of VWF strings on endothelial surfaces are tightly regulated by ADAMTS13 through proteolytic cleavage of VWF strings. The length and thickness of VWF multimers strongly correlate with physiological hemostatic potential. Therefore, the cleavage or reduction of VWF multimers by ADAMTS13 is critical for maintaining normal hemostasis.7
- vWF release in response to agonists such as:
- In plasma, VWF is present as a series of repeating subunits, ranging in size from ~500 kDa to ~20,000 kDa.8
- Basal secretion:
- Function of circulating vWF:
- vWF recruits platelets to sites of vascular injury (bridges platelets to sites of vascular damage):
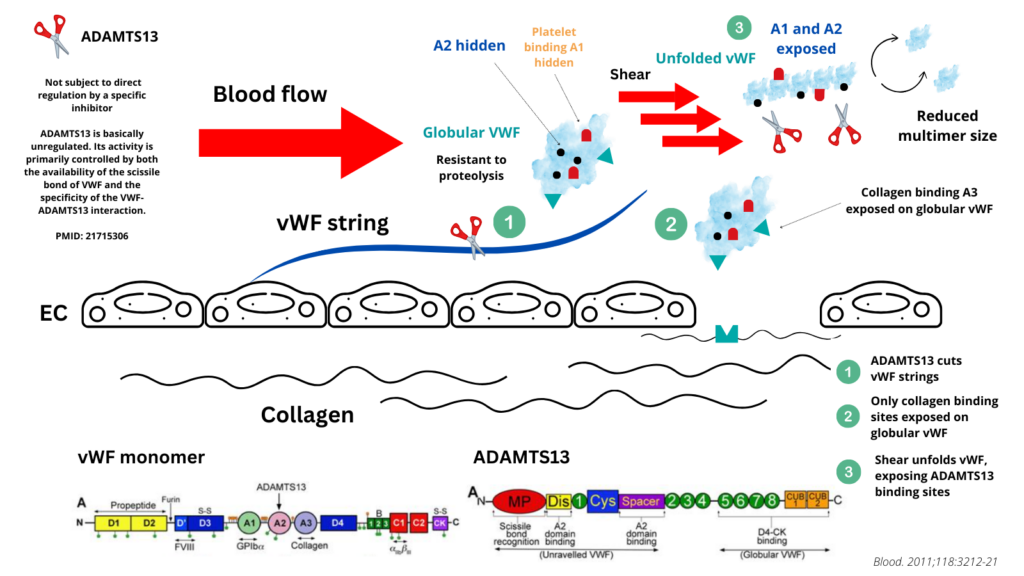
- vWF multimers transform from a globular circulating inactive conformation into an elongated, active form upon binding to the subendothelial collagen matrix via the VWF-A1 and VWF-A3 domains.9 The elongated, “active” conformation exposes previously hidden platelet binding sites that mediate the capture of circulating platelets to the site of vascular injury.
- Once unfolded, vWF-mediated platelet adhesion to collagen and aggregation are promoted via interactions between the VWF-A1 domain with the platelet glycoprotein Ib-IX-V complex and the VWF C4-domain RGD-motif with platelet integrin αIIbβ3.
- The largest multimers being the most hemostatically active.
- This same process of shear-dependent unfolding that activates VWF into a functional hemostatic protein also represents a primary determinant of ADAMTS13-medaited proteolytic regulation that controls VWF function.10
- vWF acts a carrier molecule for coagulation FVIII, which binds with high affinity to the VWF D’–D3 region. This interaction prevents FVIII from accelerated clearance and proteolysis.
- vWF recruits platelets to sites of vascular injury (bridges platelets to sites of vascular damage):

- ADAMTS13 binding to vWF (see below for more details):
- Once unfolded, the A2 domain of vWF contains an extended interactive site for several domains that are contained within ADAMTS13.
- Proteolytic regulation of vWF multimeric size occurs via hydrolysis of the Tyr1605-Met1606 peptide bond in the A2 domain by the metalloprotease ADAMTS13.
- In the absence of ADAMTS13 activity, as seen in patients with ADAMTS13 mutations or acquired autoantibodies that block plasma ADAMTS13 activity, endothelium-anchored ULVWF strings are capable of recruiting flowing platelets and causing uncontrolled thrombosis in terminal arterioles and capillaries.11
ADAMTS13
- Nomenclature:
- ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin type repeats, 13)
- History:
- First purified by Furlan and Tsai in 1996. The protease did not cleave VWF unless the substrate was subjected to fluid shear stress or treated with low concentrations of protein denaturants such as urea or guanidine hydrochloride. 12
- In 2001, several groups independently isolated VWF cleaving-protease (VWF-cp) from plasma and determined its partial amino acid sequence. This led to the identification of VWF-cp as a novel member of the ADAMTS family of metalloproteases.
- The gene encoding VWF-cp was designated ADAMTS13 according to the HUGO Gene Nomenclature Committee.
- ADAMTS13 gene:
- 29 exons panning 37 kb in the genomic sequence at chromosome location 9q34.2.
- Encodes a plasma metalloprotease that cleaves ultra large vWF.
- Expression of ADAMTS13:
- ADAMTS13 is synthesized primarily in hepatic stellate cells, which reside in the interstitial space between hepatocytes.13
- Also produced in trace amounts in:1415
- Endothelial cells
- Megakaryocytes and platelets
- Renal podocytes
- Tubular epithelial cells
- Glial cells
- Plasma concentrations of ADAMTS13 are regulated at the transcriptional and posttranslational levels under diverse (patho)physiological conditions.16
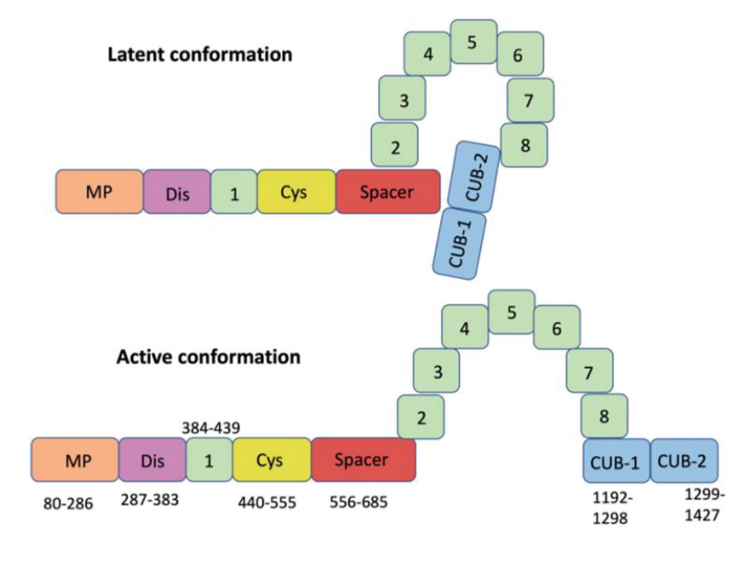
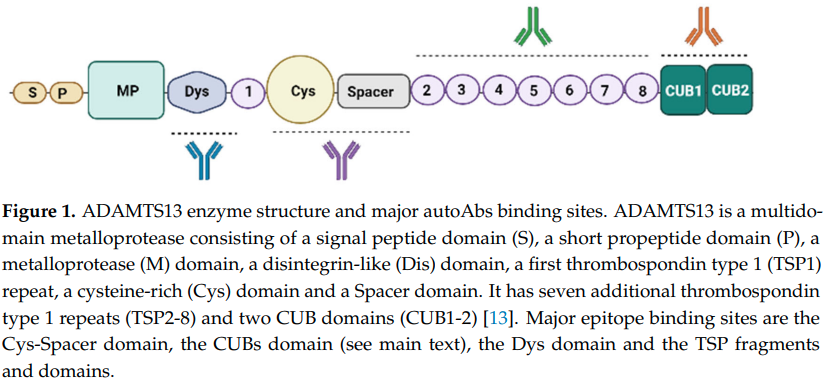
- ADAMTS13 protein:17
- Primary translation product consists of 1,427 amino acid residues.
- Composed of multiple domains (in the following order from N to C terminus):
- An N-terminal reprolysin-type metalloprotease domain (M)
- A disintegrin domain (D)
- A thrombospondin-1–like domain (T)
- A cysteine-rich domain (C) that contains an arginine-glycine-aspartate sequence
- A spacer domain (S)
- 7 additional thrombospondin-1-like domains (T2-8)
- 2 nonidentical CUB-type domains (CUB1-2)
- The physiologic functions of most of the domains are unknown.
- ADAMTS13 has a plasma half-life of 2 to 3 days.18
- The concentrations of ADAMTS13 in human plasma are 0.7–1.4 μg/ml.19
- 3% to 5% of ADAMTS13 circulates bound to vWF.20
- ADAMTS13 function:21
- ADAMTS13 is a VWF cleaving protease (VWF-cp).
- Even though ADAMTS13 is released by hepatic stellate cells as an active enzyme, ADAMTS13 circulates in a “closed” conformation through the interaction between CUB and spacer domains, preventing it from proteolyzing unselected substrates and reducing susceptibility to inhibitors (in contrast, it is found in open conformation during the acute episode of iTTP).22
- vWF proteolysis by ADAMTS13 is regulated by conformational changes in the VWF substrate rather than by activation/inhibition of the enzyme.2324
- Under conditions of shear stress, vWF unravels from a resting globular state (the latter is essentially resistant to proteolysis by ADAMTS13) to expose A1 and A2 domains, hosting platelet and ADAMTS13 binding sites respectively.25
- Plasma ADAMTS13 cleaves multimeric VWF at the Tyr1605-Met1606 bond in the central A2 domain.
- Proteolytic cleavage is shear dependent; fluid shear stress, found in the microcirculation, promotes unfolding of vWF resulting in exposure of its A2 domain for ADAMTS13 binding.26
- Exposure of the ADAMTS13 binding sites allows for ADAMTS13 binding, resulting in proteolysis of vWF (cleavage of UL-VWF results in smaller VWF multimers that are not prothrombotic).27
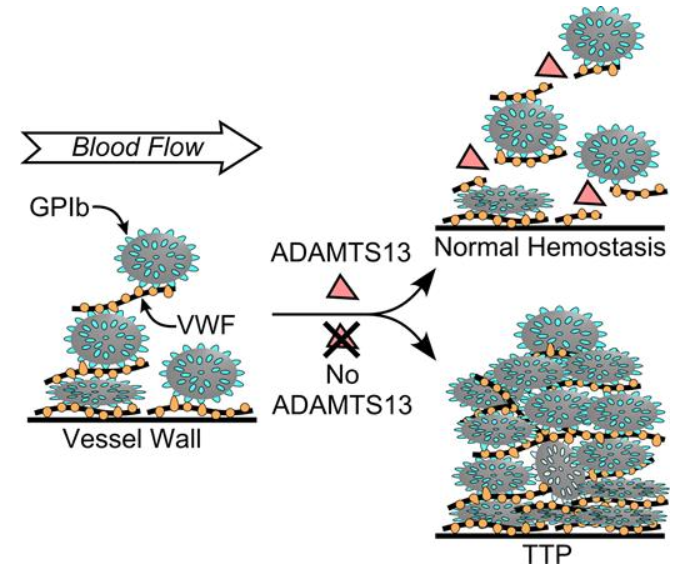
- When ADAMTS13 is deficient, UL-VWF multimers have the potential to accumulate in plasma, which can cause spontaneous platelet binding and agglutination. This can, in turn, precipitate TTP, a serious microangiopathic disorder in which platelet-rich microthrombi block arterioles and capillaries, leading to organ failure and death, if left untreated.


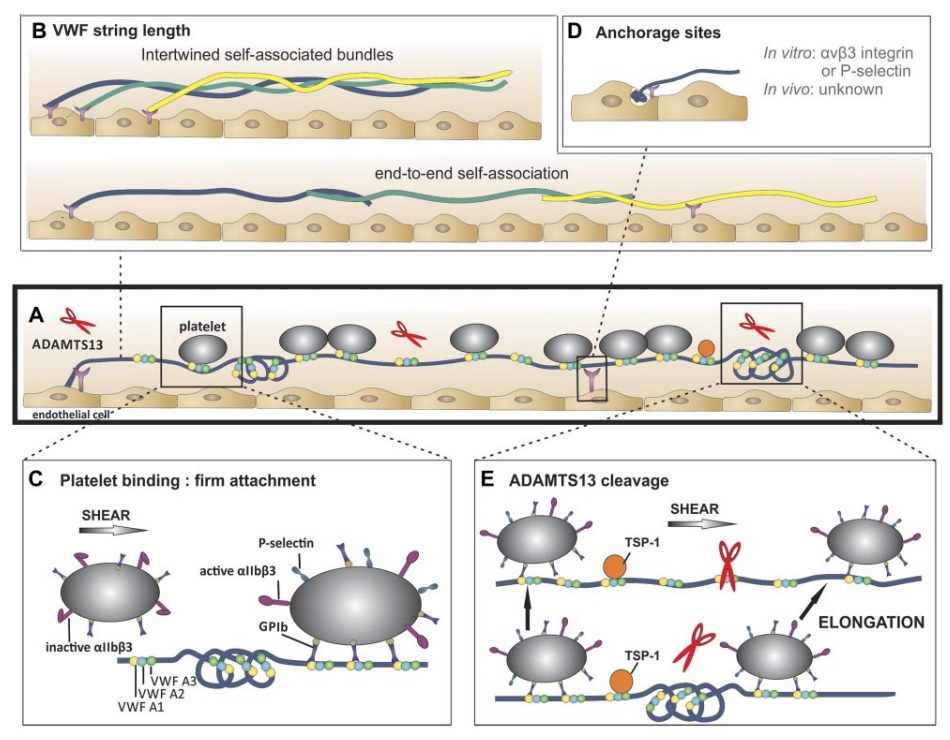
multimers. Bottom: Occasionally, extraordinary long VWF strings (> 1 mm) are observed. Hence, these strings are probably formed by end-to-end self-association of different VWF multimers. (C) Platelet binding to VWF strings. Platelets bind to open VWF A1 domains (yellow circles) via their GPIb receptors. This binding does not result in a rolling movement of the platelets but in a firm adhesion to VWF strings. Bound platelets become activated as evidenced by the presence of P-selectin and activated aIIbbetaIII on their surface. (D) Anchorage of VWF strings to endothelial cells. VWF strings are expelled from the WPB and remain anchored to the endothelial cells. P-selectin and avb3 have been identified as possible receptors involved in VWF string attachment in vitro, whereas these receptors do not seem to play a role in vivo. (E) ADAMTS13-mediated proteolysis of VWF strings. Top: ADAMTS13 (red scissors) mediated proteolysis of VWF strings is regulated by conformational changes in VWF and unfolding of its A2 domain (blue circle) induced by shear stress. Proteolysis occurs preferentially at sites of local elongations of VWF strings, which can be monitored by an increase in interplatelet distances (arrows).43 Bottom: ADAMTS13 cannot access the cleavage site in VWF A2 domains (blue circle) that are folded or cannot bind when TSP-1 (orange circle) interacts with the VWF A2-A3 domains. Source.
ADAMTS13 in TTP
- ADAMTS13 activity <10% is pathognomonic of TTP.
- TTP results from a severe deficiency of ADAMTS13 which is responsible for impaired proteolytic processing of high-molecular-weight von Willebrand factor (HMW-VWF) multimers that, under shear stress of >30 dyn/cm2 , are stretched and form long strings that are able to avidly interact with platelets and subendothelial collagen.28
- Severe deficiency of ADAMTS-13, either congenital or acquired, can result in an excess of ULVWF and cause TTP.
- Reduced ADAMTS13 activity in TTP is caused by:
- Genetic mutations, leading to hereditary TTP:
- Patients with congenital TTP (cTTP) have severe ADAMTS13 deficiency due to bialelic mutations in the coding gene (autosomal recessive)
- Mutations are found throughout all domains of the ADAMTS13 gene, including missense, frameshift, insertions, deletions and splice site alterations.29
- Inhibitory autoantibodies, leading to immune-mediated (iTTP):
- Such antibodies counteract the ADAMTS13 function, leaving VWF polymers uncleaved.
- The autoimmune response against ADAMTS13 is polyclonal and heterogeneous.30
- ADAMTS13 autoantibodies are primarily composed of immunoglobulin G (IgG), approximately 90% of which are of the IgG4 subtype.31
- Antibodies may:32
- Reduce activity of ADAMTS13:
- These are termed neutralizing or inhibiting antibodies.
- They block the proteolytic activity of ADAMTS13 towards vWF.
- Reduce circulating antigen (non-neutralizing antibodies), for example by:
- Increasing ADAMTS13 clearance (“clearing” antibodies).
- Interfering with ADAMTS13 interaction with cells or other plasma proteins.
- Reduce activity of ADAMTS13:
- Genetic mutations, leading to hereditary TTP:
Want to explore this further?
Check out the related sections in our TTP module:
- Quiz on TTP Pathophysiology
- Video lecture on TTP Pathophysiology