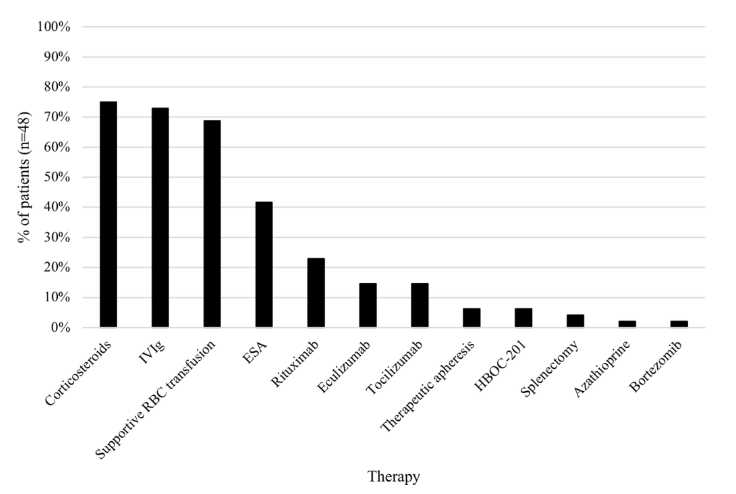
I posted a poll on twitter asking how to treat a patient with an acute complication of sickle cell disease (SCD) presenting with the data shown below:
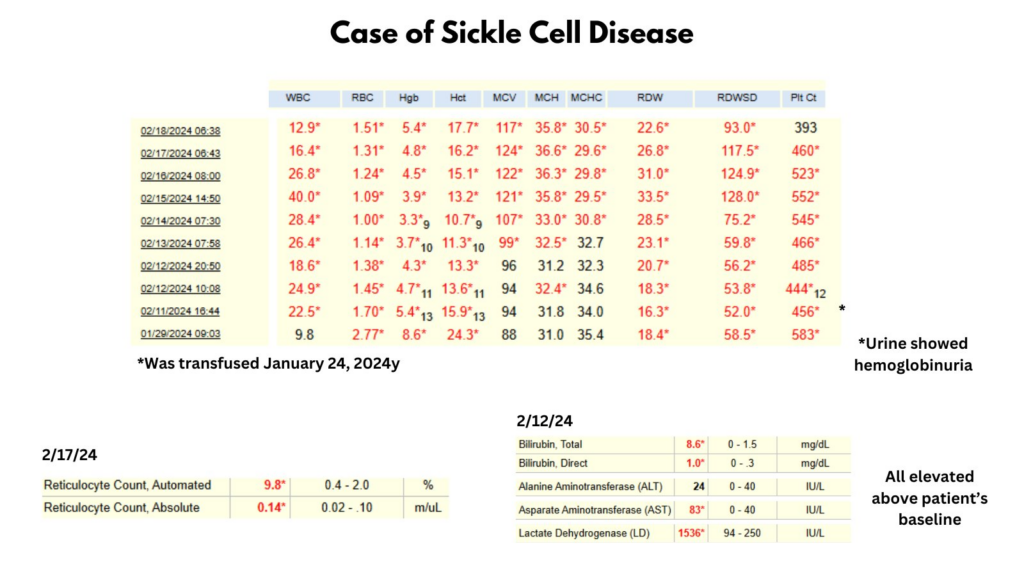
The most popular answer was red cell tansfusions:
The correct answer was IVIG!
Let’s approach the case from the standpoint of acute anemia. Acute anemia in a patient with SCD is defined as a hemoglobin (Hb) decrease ≥ 2 g/dL (20 g/L) below baseline. This patient’s Hb of 3-4 clearly qualifies as acute anemia. Causes of acute anemia include:
- Acute splenic sequestration
- Acute haptic sequestration
- Aplastic crisis (Parvovirus B19)
- Increased hemolysis for:
- Pain crisis
- Acute chest syndrome
- Acute infection
- Delayed transfusion reaction
- Acute blood loss
In this case, the severe anemia is accompanied by elevated hemolytic markers, including LDH, AST and bilirubin. Thus, the cause of the acute anemia appears to be hemolysis. Pain crisis and acute chest syndrome are typically associated with a reduction in Hb of about 1 g/dL. Likewise, with rare exceptions, acute infection would not be expected to lead to this degree of severe hemolysis. That leaves delayed transfusion reaction, more specifically hyperhemolysis syndrome) as a likely possibility. What is the evidence?
- The patient was recently transfuse.
- The Hb presumably dips below pretransfusion level (a safe assumption given the extreme degree of anemia) because both donor (allogeneic) and recipient (autologous) red cells are destroyed.
- While the reticulocyte count is by definition “appropriate” because it exceeds 0.12 x 1012/L, it is less than one would expect for this degree of anemia, suggesting some element of suppression of erythropoiesis.
In short, this is a case of hyperhemolysis syndrome in a patient with SCD. As we will discuss below, red cell transfusions (the most popular answer in the poll) are relatively contraindicated in hyperhemolysis syndrome because they promote additional hemolysis, which may lead to multiorgan failure. First line treatment for hyperhemolysis syndrome is high dose corticosteroids and IVIG.

Case History
27 yo F with HbSS admitted to outside hospital with chest pain, back pain and fever, concern for pain crisis/acute chest syndrome. Hb 6.2 g/dL. Received 3 units of pRBC. CTA negative for pulmonary embolus but patchy consolidative opacities in the bilateral lower lobes, concerning for acute chest syndrome +/- multifocal pneumonia. She was initially started on ceftriaxone/azithromycin, but was broadened to vancomycin/ceftriaxone/azithromycin in setting of ongoing fevers. Transferred to a tertiary care center for consideration of exchange transfusion. At tertiary care hospital, exchange transfusion was deferred, She was treated with antibiotics, supplemental oxygen and folic acid 5 mg daily. Patient discharged on day 10.
12 days later (now day 22 after initial transfer to tertiary care center), she presented with hematuria and flank pain and was found to have anemia to 5.8 g/dL along with fever and leukocytosis but no evidence of GU infection or stone. Treated with antibiotics empirically. Blood on urine disptick, but no RBCs on microscopy. She received 1u pRBCs, however was unmatched for antigen genotypes. Hb subsequently decreased further to 4.7, with worsening elevation of hemolysis labs c/f hyperhemolysis. She was treated with 0.4mg IVIG 3-4 days and 100 mg IV methylprednisolone daily for hyperhemolysis. Hematology note: “Recommend deferring further transfusions while concern for hyperhemolysis still active… There is concern that any transfusion, even matched, could worsen hemolysis.” 3 days later, her Hb continued to drop and her hemolytic indices continued to rise. She was started on eculizumab 900 mg (she received meningococcal vaccine the same day and she was ordered to receive 2 weeks of penicillin prophylaxis (500mg BID) due to increased risk of N. meningitides. Her Hb increased over the next 6 days to 7.5 g/dL and her hemolytic markers improved. She was discharged home.


Introduction
- Hyperhemolysis syndrome (HHS), defined as severe (potentially fatal) hemolysis causing the hemoglobin to drop rapidly below pretransfusion levels, is considered to be the most severe form of delayed hemolytic transfusion reaction, in which there is clearance of the patient’s own red cells (bystander hemolysis) in addition to transfused cells (alloimmunization via antibodies against RBCs), leading to a fall in hemoglobin below the pretransfusion level.1
- Unlike typical delayed hemolytic transfusion reactions, immunohematologic evaluation often does not detect the presence of new RBC alloantibodies or incompatibility.2
- There is often a paradoxical decrease in erythropoiesis as reflected by reticulocytopenia.3
- HHS often involves severe, life-threatening anaemia with hemoglobin levels below 4 g/dL and can be fatal.
Definitions
- Delayed hemolytic transfusion reaction (DHTR):
- American Society of Hematology (ASH) definition: DHTR is defined as a significant drop in hemoglobin within 21 days posttransfusion associated with 1 or more of the following (and exclusion of an alternative cause):4
- New red cell alloantibody
- Hemoglobinuria
- Accelerated HbS% increase with a concomitant fall in HbA posttransfusion
- Relative reticulocytopenia or reticulocytosis from baseline
- Significant LDH rise from baseline
- CDC National Healthcare Safety Network Hemovigilance criteria: DHTR is defined as a positive DAT for antibodies developed between 24 h and 28 days after transfusion, along with a positive eluate or a newly-identified antibody, and an inadequate rise of post-transfusion hemoglobin or a rapid drop in hemoglobin to pre-transfusion levels, or unexplained spherocytes on peripheral smear.
- American Society of Hematology (ASH) definition: DHTR is defined as a significant drop in hemoglobin within 21 days posttransfusion associated with 1 or more of the following (and exclusion of an alternative cause):4
- Hyperhemolysis syndrome (HHS):
- American Society of Hematology (ASH) definition: Hyperhemolysis is defined as a rapid hemoglobin decline to below the pretransfusion level and rapid decline of the posttransfusion HbA level.5
- Considered to be the most severe form of DHTR.[/efn_note]PMID: 37074146[/efn_note]
- The term hyper in hyperhemolysis refers to the fact that there is destruction not only of the red blood cells transfused but also of autologous red blood cells.
- Bystander hemolysis is defined as the lysis of antigen-negative red blood cells. Several clinical entities are complicated by bystander hemolysis: passenger lymphocyte syndrome, post-transfusion hemolysis, delayed hemolytic transfusion reactions (DHTRs), paroxysmal nocturnal hemoglobinuria, and hemolysis complicating infectious diseases.6
Epidemiology
- Most common in patients who receive frequent transfusions, especially those with sickle cell disease (SCD) or transfusion-dependent thalassemia.7
- However, can present in any patient undergoing RBC transfusion.
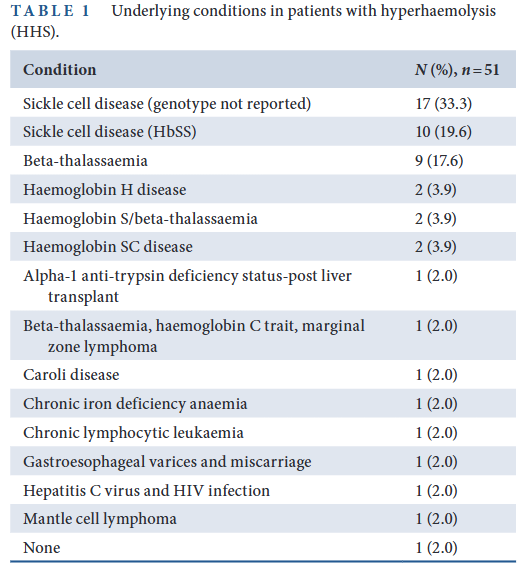
- In a systematic review of 44 studies comprising 51 patients with HHS:8
- Significantly more females than males.
- The median age of patients was 33 years (range: 1–80 years).
- Six patients developed hyperhaemolysis during pregnancy, with individuals ranging from 11 to 24 weeks of gestational age when the reaction occurred.
- Among the 51 patients with HHS, 43 (84.3%) had an underlying haemoglobinopathy and/or thalassemia, including 31 with SCD (HbSS, HbSC and HbS/β-thalassemia)
Pathophysiology
- The pathophysiology of delayed hemolytic transfusion reaction (DHTR) involves a recipient’s production of alloantibodies against a donor’s RBC antigens following a transfusion with re-exposure to the same RBC antigen triggering an anamnestic antibody response resulting in complement activation and hemolysis of allogeneic RBCs (re-stimulation of an alloantibody).9
- The pathophysiology of hyperhemolysis syndrome (HHS) is characterized by destruction of both allogeneic and autologous RBCs, with hemoglobin levels falling below the level prior to the transfusion. The pathogenesis is unclear, but several theories have been proposed, including:
- Antibody-dependent hemolysis
- Complement-mediated hemolysis
- Macrophage activation
- Bystander hemolysis involves the destruction of patient (autologous) red cells and may be mediated by:10
- Complement activation:
- Free hemoglobin can activate complement, leading to deposition of C3, which has previously been shown to opsonize cells for clearance by macrophages.
- Free heme and free Hb during DHTR promote intravascular oxidative stress, leading to abnormal activation of the alternative complement pathway.11
- Activation of both classical and alternative pathways has been identified in patients with HHS, while the C5b-9 formulation in bystander RBCs membrane is crucial for cell lysis. Intravascular hemolysis leads to the activation of alternative complement pathway and a vicious cycle is established.
- either via alloantibody formation or alloantibody-independent processes.
- A disordered hyperactive immune response
- Transiently auto-reactive antibodies
- Complement activation:
- Reticulocytopenia is a well-documented manifestation of HHS, and may be secondary to:12
- Myelosuppression
- Peripheral destruction by macrophages
- Bystander hemolysis
- Patients present with a more severe anemia than was present prior to transfusion owing to:
- Patient’s own red blood cells being destroyed
- Reticulocytopenia
Clinical Presentation
- Patients with hyperhemolysis syndrome (HHS) typically present with significant anemia (drop from pre-transfusion level) with a median hemoglobin nadir of 3.9 g/dL occurring a median of 10 days following the last RBC transfusion.13
- Patients should have a history of recent RBC transfusion usually within a week but almost always within the past 21 days.
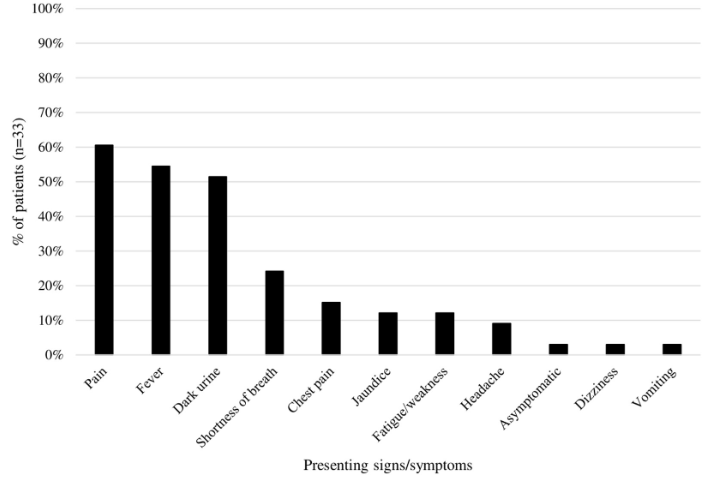
- HHS typically presents with:14
- Fever
- Jaundice
- Signs and symptoms of hemolytic anemia
- Severe pain usually out of proportion to patient’s known vasoocclusive pain
- Dark urine
- Hemolysis
- Hemoglobinuria
- Reticulocytopenia (thus, non-regenerative anemia with a lower hemoglobin level than was present pre-transfusion)
Diagnosis
- Suspect diagnosis if rapid Hb drop to below the pretransfusion level.15
- There are no published diagnostic criteria that define HHS for research purposes at an international level. The primary diagnostic criteria are:16
- Recent transfusion
- Drop in Hb
- Laboratory evidence of hemolysis
- Labs include:17
- Complete blood count
- A Hb drop greater than 25% suggests DHTR
- Hb nadir of 3.9 g/dL (inter-quartile range: 3.2–5.1; range 1.8–7.6 g/dL) with a median time to nadir of 10 days (range 1–18 days).18
- Many cases are accompanied by reticulocytopenia.19
- Increased LDH
- Among 41patients with LDH values reported, the median peak was1683 U/L (IQR: 1194–3000 U/L; range: 612–16 156 U/L).20
- Increased plasma free Hb
- Increased bilirubin
- Immunohematology labs
- Triggering alloantibody is often not present
- About 70% of patients will have positive indirect anti-human globulin testing (IAT)
- About 50% of patients have a negative direct anti-human globulin test (DAT)
- Complete blood count
- American Society of Hematology (ASH) advises serial monitoring of the hemoglobin, hematocrit, quantification of HbA and HbS fractions, reticulocyte count, bilirubin, LDH, and urinalysis (for hemoglobinuria).
Differential Diagnosis
- Acute chest syndrome or vaso-occlusive crisis, which may be accompanied by a drop in Hb from baseline.
- Typical delayed hemolytic transfusion reaction (DHTR).
Treatment
- Many cases are managed with multiple therapies to maximize the likelihood of survival.
- Avoid further transfusion (unless hemodynamic status becomes critical) to prevent worsening hemolysis, multiorgan failure and death:21
- Paradoxically, management usually involves avoidance of transfusion since additional allogeneic RBCs are thought to potentiate subsequent episodes of hemolysis.
- Supportive RBC transfusion may be associated with a significantly longer time to recovery compared to patients that did not receive additional transfusion.
- Prior reports have shown that patients with HHS may experience rapid decompensation, particularly following RBC transfusion, which can be fatal.
- For patients experiencing life-threatening anemia, transfusion with extended matched red cells that also lack the offending antigen should be considered.
- ASH: Avoidance of further transfusion is recommended unless patients are experiencing life-threatening anemia with ongoing hemolysis. If transfusion is warranted, extended matched red cells (C/c, E/e, K, Jka/Jkb, Fya/Fyb, S/s) should be considered.
- Measures to prevent iatrogenic anemia:22
- Limiting blood draws
- Eliminating unnecessary laboratories
- Using pediatric tubes for testing
- Optimize erythropoiesis:23
- Assess for nutritional deficiency, including iron, vitamin B12, and folate levels, although empiric use may be considered depending on the patient population.
- Medications that can lead to marrow suppression, such as hydroxyurea, should be discontinued.
- Erythroid stimulating agents (ESA) (with or without IV iron) should be considered at high doses to help drive RBC production.
- Dampen the immune response:24
- Steroids
- IVIG
- Complement inhibitors
- C5a with eculizumab
- Interleukin-6 (IL-6) blockers
- American Society of Hematology (ASH) recommendations:
- The ASH guideline panel suggests immunosuppressive therapy (IVIG, steroids, rituximab, and/or eculizumab) over no immunosuppressive therapy in patients with SCD (all genotypes) with a delayed hemolytic transfusion reaction and ongoing hyperhemolysis (conditional recommendation based on very low certainty in the evidence about effects.
- First-line immunosuppressive agents include:
- High-dose steroids (methylprednisolone or prednisone at 1 to 4 mg/kg per day)
- IVIG (t 0.4 to 1 g/kg per day for 3 to 5 days (up to a total dose of 2 g/kg))
- Second-line agent is eculizumab (900 to 1200 mg weekly for hyperhemolysis for patients > 40 kg) for patients who continue to experience clinical deterioration despite first-line agents. When considering eculizumab, immediate vaccination with Menveo (MenACWY), either Bexsero or Trumenba (MenB), and ciprofloxacin prophylaxis are advised to reduce the risk for meningococcal infection
- Rituximab is primarily indicated for potential prevention of additional alloantibody formation in patients who may require further transfusion.
- If a patient is experiencing life-threatening anemia, transfusion should not be withheld, and if feasible, extended antigen matched red cells (C/c, E/e, K, Jka/Jkb, Fya/Fyb, S/s) should be transfused.