Introduction
Thalassemia refers to a group of inherited disorders of hemoglobin synthesis caused by reduced or absent production of globin chains. The imbalance between α- and β-globin chains leads to ineffective erythropoiesis, hemolysis, and varying degrees of anemia. Thalassemia is broadly classified by which globin chain is affected (α vs β), the underlying genetic lesion, and the clinical severity of the disease. Understanding the classification is essential for diagnosis, prognosis, and management.
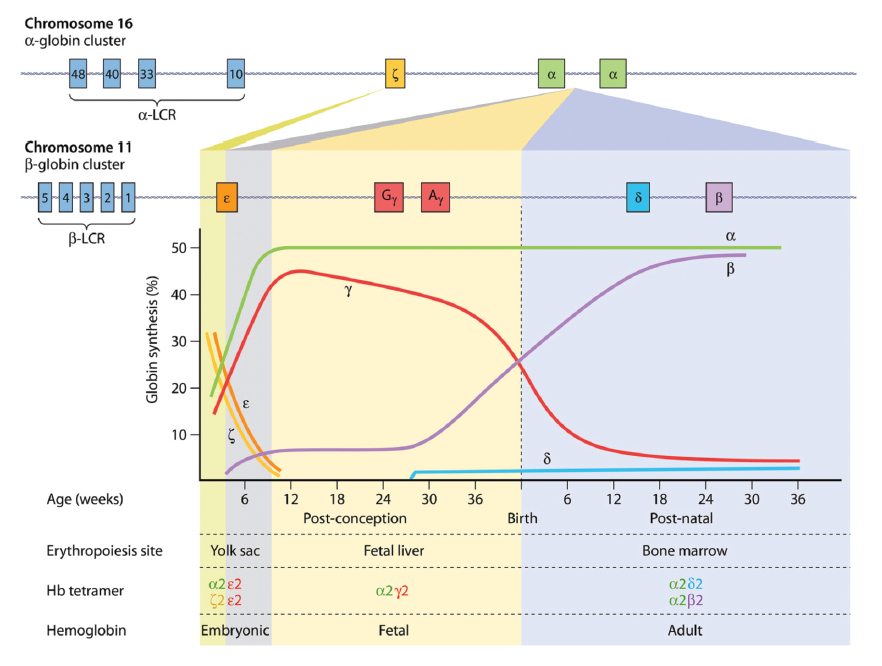
expressed from chromosome 16 pair with two monomers of β-like globin to form a functional
haemoglobin tetramer. The components of the haemoglobin change during development. The
embryonic (ε) part of β-like globin is expressed during the first 6 weeks of gestation and forms embryonic
haemoglobin by pairing with α-globin. After 6 weeks, ε-globin expression declines and is replaced by γ-
globin to form foetal haemoglobin. The final haemoglobin switching takes place a few weeks before
birth when the γ-globin expression is replaced by β-globin to form adult hemoglobin. Abbreviations:
Hb, hemoglobin; LCR, locus control region.
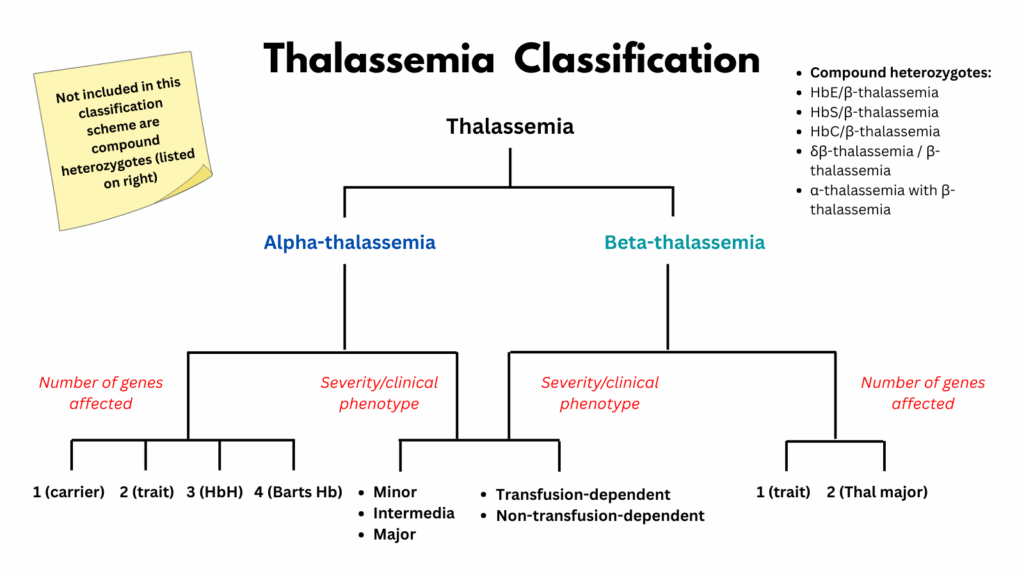
Classification by Globin Chain Affected
Normal hemoglobin consists of two α-globin chains and two β-globin chains. In normal individuals, there are four functional α-globin genes, with two inherited from each parent (αα/αα), and two β-globin genes, with one inherited from each parent (β/β).
- α-Thalassemia:
- Reduced or absent synthesis of α-globin chains
- Most often due to deletions in the α-globin genes (on chromosome 16)
- Deletions that result in the loss of duplicated α-genes lead to the absence of α-globin production from that chromosome, and they are called α0-thalassaemia deletions (–), and those that result in the loss of single a gene and decreased production of α-globin from that chromosome are called α+-thalassemia deletions (-α)
- Less frequently, α-thalassemia can occur due to point mutations in either HBA1 or HBA2, resulting in the production of abnormal or unstable variant α-globin chains (αTα or ααT) (non-deletional α+-thalassaemias (αTα or ααT).
- There are 2 clinically significant forms of α-thalassemia:
- Hemoglobin (Hb) Bart hydrops fetalis syndrome (α-thalassemia major)
- Hemoglobin H (HbH) disease (α-thalassemia intermedia)
- β-Thalassemia:
- Reduced or absent synthesis of β-globin chains
- Usually due to point mutations in the β-globin gene (on chromosome 11)
Classification by Genotype
- α-thalassemia:
- The clinical spectrum of α-thalassemia depends on how many of the four α-globin genes are deleted or inactivated. The severity ranges from silent carrier status to lethal hydrops fetalis, with intermediate phenotypes in between.
- Silent carrier (−α/αα); One mutated gene, asymptomatic, normal CBC
- Trait (minor) (−α/−α or −−/αα); Two mutated genes, mild microcytic anemia1
- Hemoglobin H disease (−−/−α); Three mutated genes, moderate to severe hemolytic anemia2
- Hemoglobin Bart’s Hydrops fetalis (−−/−−); Four mutated genes, lethal in utero without intervention (Hb Bart’s, γ4 tetramers)
- The clinical spectrum of α-thalassemia depends on how many of the four α-globin genes are deleted or inactivated. The severity ranges from silent carrier status to lethal hydrops fetalis, with intermediate phenotypes in between.
- β-thalassemia:
- The phenotype of β-thalassemia depends on the number and type of mutations affecting the two β-globin genes. Clinical severity ranges from asymptomatic carriers (trait) to transfusion-dependent thalassemia major, with β-thalassemia intermedia falling in between.
- Heterozygotes
- Trait (minor); usually asymptomatic, mild microcytosis
- Homozygotes or compound heterozygotes for β-thalassemia alleles
- Intermedia; variable severity, moderate anemia, may need intermittent transfusions
- Major (Cooley’s anemia, Mediterranean anemia, homozygous or compound heterozygous severe mutations), transfusion-dependent, severe anemia, bone changes, iron overload without chelation
- Heterozygotes
- The phenotype of β-thalassemia depends on the number and type of mutations affecting the two β-globin genes. Clinical severity ranges from asymptomatic carriers (trait) to transfusion-dependent thalassemia major, with β-thalassemia intermedia falling in between.
Classification by Transfusion Requirement
- Thalassemia can also be classified by transfusion requirement. Patients with transfusion-dependent thalassemia (TDT) require regular lifelong transfusions for survival, while those with non–transfusion-dependent thalassemia (NTDT) generally maintain hemoglobin without regular transfusions but may need support during stress or complications.
- Transfusion-Dependent Thalassemia (TDT):
- Regular, lifelong transfusions required for survival
- Non–Transfusion-Dependent Thalassemia (NTDT):
- Do not require regular transfusions for survival, but may need occasional support during stress (infection, pregnancy, surgery)
- Transfusion-Dependent Thalassemia (TDT):
Classification by Clinical Severity (‘conventional phenotypes’)
- Thalassemias may also be classified as “trait,” “minor,” “intermedia,” and “major,” reflecting a spectrum of severity, with trait/minor being the mildest and major the most severe.
- Thalassemia minor/trait: usually clinically silent, mild anemia
- Thalassemia intermedia: moderate anemia, not always transfusion-dependent
- Thalassemia major: severe anemia, transfusion-dependent
| Clinical Category | α thalassemia | β thalassemia |
|---|---|---|
| Minor/trait | α thalassemia trait | β thalassemia minor |
| Intermedia | Hb H disease | β thalassemia intermedia |
| Major | Hydrops fetalis | β thalassemia major (Cooley’s) |
Special Categories
- δβ-Thalassemia: deletions affecting both δ and β genes, ↑ HbF
- δβ-thalassemia is a rare inherited disorder caused by deletions in the β-globin gene cluster on chromosome 11 that involve both the δ-globin (HBD) and β-globin (HBB) genes.
- This results in absent or severely reduced production of both δ- and β-globin chains.
- In compensation, γ-globin chain production persists, leading to elevated levels of fetal hemoglobin (HbF, α₂γ₂).
- Most cases are due to large deletions removing both δ- and β-globin genes.
- These deletions may vary in size and location, but all impair the normal switch from γ-globin to β-globin expression after birth.
- Unlike HPFH, where γ-globin expression is upregulated without anemia, δβ-thalassemia has a thalassemic phenotype.
- Hereditary persistence of fetal hemoglobin (HPFH): benign persistence of HbF into adulthood
- HPFH is a benign genetic condition characterized by continued production of fetal hemoglobin (HbF, α₂γ₂) into adulthood.
- It results from deletions or point mutations in the β-globin gene cluster on chromosome 11 that interfere with the normal switching off of γ-globin gene expression after birth.
- HPFH is not classified as a type of thalassemia.
- Thalassemias are disorders of globin chain synthesis (quantitative defects), while HPFH involves persistent expression of the γ-globin chains.
- However, HPFH can mimic thalassemia and may be confused with β-thalassemia trait due to:
- Microcytosis
- Elevated HbF levels
- Normal HbA₂
- Compound heterozygous states: e.g., HbE/β-thalassemia, HbS/β-thalassemia — clinically important, variable severity
- Hb E disease
- Hemoglobin E is a structural hemoglobin variant caused by a G→A mutation at codon 26 of the β-globin gene (HBB).
- This mutation leads to a glutamic acid → lysine substitution and simultaneously activates a cryptic splice site, resulting in reduced β-globin synthesis.
- Although HbE is a structural variant, it behaves functionally like a mild β⁺-thalassemia:
- Abnormal hemoglobin structure
- Reduced β-globin production
- Therefore, HbE is often classified as a thalassemic hemoglobinopathy, especially when inherited with other β-globin mutations.
- Unusual forms of α-thalassemia:
- ATR-16 Syndrome
- Rare genetic disorder caused by large deletions at the telomeric end of chromosome 16, affecting the α-globin gene cluster
- Characterized by a triad of:
- α-thalassemia
- Cognitive impairment
- Dysmorphic facial features
- Results from contiguous gene deletion beyond just the α-globin genes
- ATR-X Syndrome
- X-linked genetic disorder caused by mutations or deletions in the ATRX gene on the X chromosome
- Primarily affects boys, with features including:
- Severe intellectual disability
- α-thalassemia
- Recognizable craniofacial dysmorphism
- ATRX gene encodes a protein that regulates chromatin remodeling
- Plays a role in incorporating histone variant H3.3 into telomeric and pericentromeric DNA
- Mutation leads to downregulation of α-globin gene expression
- α-Thalassemia Myelodysplastic Syndrome (ATMDS)
- An acquired form of α-thalassemia seen in patients with:
- Myelodysplastic syndromes (MDS)
- Other hematologic malignancies
- Usually results from somatic mutations in the ATRX gene
- Rarely, may be due to acquired deletions of the telomeric end of chromosome 16
- Should be suspected in MDS patients with unexplained microcytic anemia
- An acquired form of α-thalassemia seen in patients with:
- ATR-16 Syndrome
Evolving Frameworks for Thalassemia Classification
Thalassemia has historically been classified by clinical severity (minor, intermedia, major). While this system is useful for describing disease biology, modern practice often relies on a more practical framework: transfusion-dependent (TDT) vs. non–transfusion-dependent (NTDT) thalassemia. Each system offers value, but they serve different purpose; one highlights phenotype, the other guides management. TDT vs NTDT is the preferred modern classification in guidelines because it directly translates into management pathways, risk profiles, and monitoring requirements. By contrast, Minor / Intermedia / Major remains valuable for teaching disease biology and natural history, but it’s less helpful for everyday clinical decision-making.
Traditional Classification: Minor / Intermedia / Major
-
Advantages:
- Long-established and widely recognized in hematology.
- Provides a spectrum of disease severity (mild → moderate → severe).
-
Useful for describing natural history and phenotype-genotype correlations.
-
Limitations:
- Terms like intermedia are vague and variable — patients with the same label may differ widely in transfusion needs.
- It does not directly guide management decisions (e.g., transfusion vs. no transfusion).
-
Less relevant in the era of individualized therapy and disease-modifying treatments.
Modern Classification: NTDT vs. TDT
-
Advantages:
- Clinically actionable — directly linked to transfusion strategy, iron chelation, and monitoring.
- Clear-cut: patients either require regular lifelong transfusions (TDT) or they do not (NTDT).
-
Better reflects real-world management and is used in most contemporary clinical trials and guidelines.
-
Limitations:
- Simplifies a continuum into two categories — some NTDT patients may drift toward transfusion dependence over time.
-
Does not capture genetic diversity or subtle differences in disease biology.
How they relate
- Thalassemia major ≈ TDT
- Thalassemia intermedia ≈ often NTDT (though some patients become TDT later)
- Thalassemia minor/trait ≈ neither TDT nor NTDT (asymptomatic carriers)
Note:
It is important to differentiate between inherited α-thalassemia (focus of this topic) from α-thalassemia-myelodysplastic syndrome (acquired form of HbH disease) which results from acquired ATRX pathogenic gene variant that occurs primarily in older male adults with myelodysplasia.
References
- Kattamis A, Kwiatkowski JL, Aydinok Y. Thalassaemia. Lancet. 2022 Jun 18;399(10343):2310-2324.
- Harteveld CL, Achour A, Arkesteijn SJG, et al. The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int J Lab Hematol. 2022 Sep;44 Suppl 1:28-36.
- Cappellini, MD, Farmakis, D, Porter, J, et al. Thalassaemia International Federation 2025 Guidelines for the Management of Transfusion Dependent Thalassaemia. 5th Edition, 2025.
- Taher et al. β-Thalassemias. N Engl J Med. 2021 Feb 25;384(8):727-743.