Postscript
Introduction:
- Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease with variable multisystem involvement and heterogeneous clinical features, ranging from mild to life threatening.
- Diagnosis is made on clinical grounds in the presence of characteristic serological abnormalities and is typically based on established classification criteria, including:
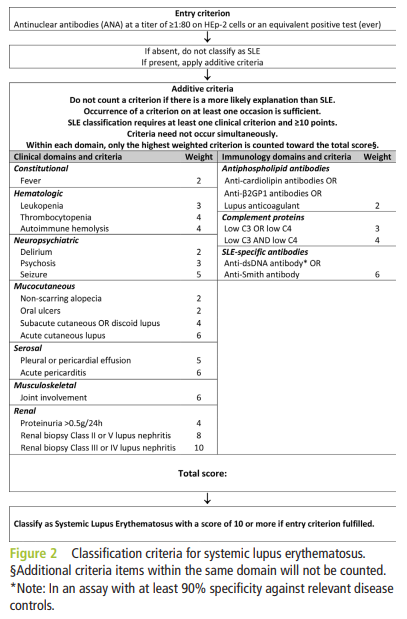

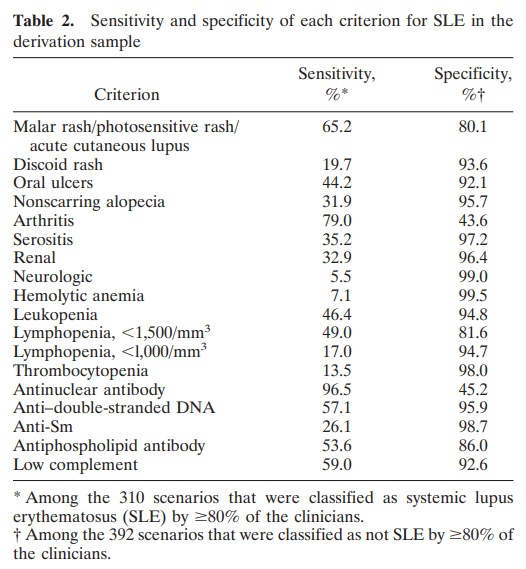
- Hematological abnormalities are common findings in patients with SLE:
- Major hematologic manifestations of SLE include:
- Anemia
- Leukopenia
- Thrombocytopenia
- Lymphadenopathy
- Splenomegaly
- Antiphospholipid syndrome
- Bone marrow may also be a target leading to such conditions as:
- Myelofibrosis
- Pure red cell aplasia
- Aplastic anaemia
- Presentation may occur:
- At the time of diagnosis:
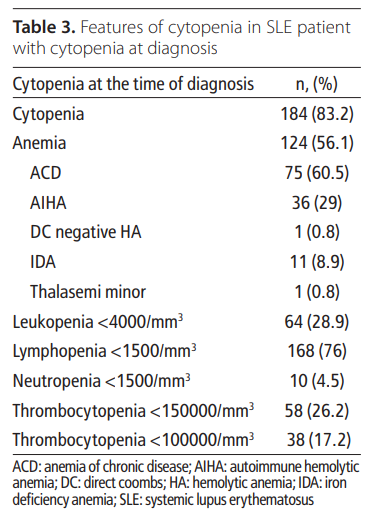
- In a cohort of 221 patient, cytopenias:
- Were present in 83.3% at diagnosis.
- Developed in up to 90% during follow-up.
- In a cohort of 221 patient, cytopenias:
- Throughout the course of the disease, especially with disease flares.
- At the time of diagnosis:
- Mechanisms of hematological changes may reflect:
- Manifestation of the autoimmune process itself:
- Most cytopenias are directly caused by autoantibodies, most of which cannot be routinely measured (with exception of positive Coombs test in autoimmune hemolytic anemia).
- Consequence of SLE treatment
- An SLE- and treatment-independent process
- Manifestation of the autoimmune process itself:
- Major hematologic manifestations of SLE include:

- Diagnostic criteria for SLE include the following hematological parameters:
- Hemolytic anaemia
- Leukopenia
- Lymphopenia
- Thrombocytopenia
- Antiphospholipid antibodies
White blood cells
Overview
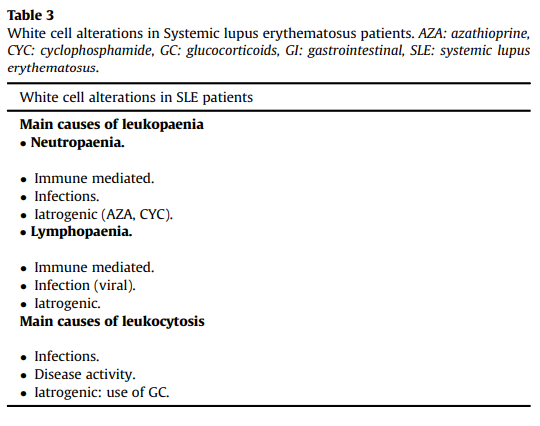
Leukopenia
Definition
- 2019 EULAR/ACR and 2012 SLICC diagnostic criteria define leukopenia as white blood cell (WBC) count < 4.0 x 109/L:
- EULAR/ACR found that leukopenia found on one occasion had a higher sensitivity and specificity than leukopenia defined on two or more occasions.
- SLICC stipulates that leukopenia should only be counted in the absence of other known causes such as Felty’s syndrome, drugs, and portal hypertension.
- Leukopenia may include one or both of:
- Neutropenia – common and usually mild
- Lymphopenia:
- The most common WBC abnormality among patients with SLE.
- Correlates with disease activity.
Epidemiology
- Overall, leukopenia (white blood cell [WBC] <1.0 x 109/L ) occurs in 50–60% of patients with SLE.
- WBC count <1.0 x 109/L in 17% of patients.
- In a cohort of 221 patients:
- 28.9% had leukopenia at the time of diagnosis.
- 48% developed leukopenia follow-up.
- In a cohort of 115 patients, 57% had leukopenia.
Treatment
- According to the 2019 EULAR guideline recommendations: autoimmune leukopenia is common in SLE but rarely needs treatment; careful work-up is recommended to exclude other causes of leukopenia (especially drug-induced).
Neutropenia
Epidemiology
- Overall, neutropenia occurs in 20–47% of patients with SLE.
- In a cohort of 126 patients with SLE, 5% had moderate to severe neutropenia (<1.0 x 109/L or < 0.5 x 109/L, respectively).
- In a cohort of 124 patients with SLE, 0.8% had absolute neutrophil count <1.0 x 109/L.
- In a cohort of 998 patients with SLE (mostly Caucasian), 21% had neutropenia.
- In a Asia Pacific Lupus Collaboration cohort of 2330 patients, 12.5% had neutropenia.
- In a cohort of 221 patients:
- 4.5% had neutropenia at the time of diagnosis.
- 10% developed neutropenia during follow-up.
- In a cohort of 115 patients, 20% had neutropenia (< 1.8 x 109/L).
- Risk factors for developing neutropenia include:
- Thrombocytopenia
- Central nervous system involvement
- Immunosuppressive medications
Pathogenesis
- Causes include:
- Underlying SLE itself (autoimmune)
- Viral infections
- Immunosuppressive medications
- Hypersplenism
- Co-incidental benign ethnic (familial) neutropenia:
- One of the most common causes of chronic neutropenia seen in individuals of African, Middle Eastern, and west Indian descent.
- Should be excluded before attributing neutropenia to SLE.
- Mechanisms of neutropenia include:
- Increased peripheral destruction of granulocytes:
- Mainly due to circulating antineutrophil antibodies, including:
- Non-complement-activating
- Complement-activating
- The specific target of the autoantibodies is unknown.
- In one study, patients with SLE with anti-Ro autoantibodies were found to have significantly lower neutrophil counts than patients with SLE without anti-Ro.
- Mainly due to circulating antineutrophil antibodies, including:
- Decreased marrow production:
- Antibodies may also target marrow precursors, resulting in bone marrow underproduction.
- T cell-mediated or monocyte-mediated suppression of bone marrow granulocytopoiesis.
- Increased levels of neutrophil apoptosis and aberrant clearance of apoptotic bodies.
- Changes in marginal and/or splenic pool.
- Increased peripheral destruction of granulocytes:
Clinical presentation
- Recurrent infections are the only known significant consequence of neutropenia.
- Local signs and symptoms of infection may be attenuated in patients with SLE due to immunosuppression.
- Moderate to severe neutropenia (ANC <1000/microL) may be accompanied by:
- Infection (either as a cause or as a consequence of the neutropenia)
- Other cytopenias including lymphopenia and thrombocytopenia
- Low complement C3, C4
- Positive Coombs test
- History of neuropsychiatric involvement
- In a Asia Pacific Lupus Collaboration cohort of 2330 patients neutropenia negatively correlated with:
- Methotrexate use
- Cyclosporine use
- Low disease activity
Treatment
- Generally requires no specific therapy.
- Recombinant granulocyte-colony stimulating factor (G-CSF) may be considered for severe and life-threatening neutropenia, but may cause disease flare.
Lymphopenia
Definition
- Original 1982 ACR criteria included lymphopenia, defined as < 1.5×109 on at least 2 occasions.
- 2019 EULAR/ACR diagnostic criteria do not include lymphopenia. Not voted into the final set of criteria by the external experts in the nominal group exercise for the EULAR/ACR criteria, owing to poor specificity.
- 2012 SLICC diagnostic criteria include lymphopenia as defined by an absolute lymphocyte count <1.5×109 /L on at least one occasion in the absence of other known causes such as corticosteroids, drugs, and infection.
- Both T and B lymphocyte subpopulations may be affected.
- Though T cell numbers (especially the CD4+ subset) are affected more than B cells, the CD4/CD8 ratio is usually unchanged.
Prevalence
- Low lymphocyte counts commonly occur in SLE with a prevalence ranging from 20% to 93%.
- Lymphocyte count <0.5×109/L observed in 10% of patients.
- In a Asia Pacific Lupus Collaboration cohort of 2330 patients, 37.2% had lymphopenia. Cumulatively lymphopenia detected during follow-up in 79.2%.
- In a cohort of 115 patients with SLE, 82% had lymphocyte count <1.5×109/L.
- Occurs independently of neutropenia.
- Lymphocyte levels may fluctuate during the clinical course, irrespective of treatment.
- Observed more frequently in patients with active or severe disease.
Pathogenesis
- Causes include:
- Underlying SLE itself (autoimmune)
- Non-SLE causes including:
- Viral infections:
- Cytomegalovirus
- Parvovirus B19
- HIV
- Medications
- Malignancy
- Viral infections:
- Mechanisms remain unclear:
- Likely involve antilymphocyte antibodies:
- Heterogenous group of autoantibodies.
- Identified in vitro by their ability to lyse lymphocytes or by their binding to the surface of lymphocytes or plasma membrane components.
- Prototypic antibodies are cold-reactive IgM antibodies that mediate complement-mediated lymphocyte toxicity.
- Circulating lymphotoxic antibodies are commonly identified in SLE, and levels correlate with lymphopenia.
- Defects in apoptosis may also play a role:
- Upregulation of fas antigen on naive peripheral T cells may render them highly susceptible to fas-mediated apoptotic cell death.
- Likely involve antilymphocyte antibodies:
Clinical presentation
- Lymphopenia may be:
- Clinically silent
- Associated with:
- Increased risk of infections, though data on the increased risk of infection are controversial.
- Active SLE (disease activity), especially:
- Fever
- Polyarthritis
- Central and peripheral nervous system disease
- Predictor of SLE flares.
- In a Asia Pacific Lupus Collaboration cohort of 2330 patients lymphopenia associated with:
- Overall disease activity
- Elevated ESR
- Use of:
- Prednisolone
- Azathioprine
- Methotrexate
- Tacrolimus
- Cyclophosphamide
- Rituximab
- In a Latin American cohort of 1437 patients, lymphopenia:
- Positively associated with:
- Younger age
- Mestizo ethnicity
- Negatively associated with treatment with:
- Antimalarials
- Azathioprine
- Positively associated with:
Treatment
- No specific treatment.
- Treatment is aimed at the underlying SLE, which can lead to improvement in lymphocyte counts.
- In rare cases with severe lymphocytopenia, it may be appropriate to use prophylactic antibiotics against organisms such as Pneumocystis jirovecii, but evidence is lacking for this approach.
Platelets
Thrombocytopenia
Definition
- According to the 2019 EULAR/ACR diagnostic criteria criteria for SLE, the definition of thrombocytopenia is a platelet count <100 ×109/L.
- According to the 2012 SLICC diagnostic criteria, thrombocytopenia is defined as a platelet count <100 ×109/L on at least one occasion in the absence of other known causes such as drugs, portal hypertension, and thrombotic thrombocytopenic purpura.
Epidemiology
- Overall, 10–15% of patients with SLE have platelet count <100 ×109/L.
- About 10% of patients have platelet counts <50 ×109/L.
- 25-50% of patients have platelet counts between 100 ×109/L and 150 ×109/L.
- In about 15% of patients, isolated thrombocytopenia precedes the diagnosis of SLE by several years.
- In a cohort of 221 patients, thrombocytopenia was present in 17.2% at time of diagnosis and in 34% during follow-up.
- In a cohort of 115 patients:
- 26% of patients had platelet count < 100×109/L.
- 40% of patients had platelet count < 150×109/L.
Pathogenesis
- Causes include:
- Immune thrombocytopenia (ITP):
- SLE is a cause of secondary ITP.
- Secondary ITP (20% of cases) is ITP due to an underlying disease; other causes of secondary ITP include:
- Antiphospholipid syndrome (APS)
- Chronic lymphocytic leukemia
- Hepatitis C
- HIV
- Common variable immune deficiency
- Drug-induced thrombocytopenia
- Antiphospholipid syndrome
- Thrombocytopenia is one of the non-criteria manifestations of primary APS (with no evidence of SLE).
- Hypersplenism; thrombocytopenia in this context:
- Is typically mild to moderate (eg, platelet count between 60 ×109/L and 150 ×109/L).
- May be accompanied by mild anemia and/or mild leukopenia.
- Thrombotic microangiopathy (TMA):
- Malignant hypertension
- Catastrophic APS (CAPS)
- Thrombotic thrombocytopenic purpura (TTP)
- SLE flare
- Immune thrombocytopenia (ITP):
- Mechanisms:
- SLE-mediated ITP is mediated by anti-platelet antibodies that result in increased peripheral destruction of platelets:
- Targets of the immune response against platelets in patients with SLE may differ from the targets seen in patients with isolated ITP.
- Thrombocytopenia in patients with SLE associated with:
- aCL antibodies
- Antiphosphatidic antibodies
- Antiphosphoserine antibodies
- Antiphosphoinositol antibodies
- Lupus anticoagulant
- The known association of thrombocytopenia with the presence of aPL antibodies may, at least in some patients, be related to a common or a cross-reactive antigen.
- Drug-induced thrombocytopenia – may be immune or nonimmune.
- TMA – platelet consumption in the setting of a thrombotic microangiopathic process.
- SLE-mediated ITP is mediated by anti-platelet antibodies that result in increased peripheral destruction of platelets:
Clinical presentation
- Presentation of isolated thrombocytopenia:
- May occur prior to the development of SLE:
- First manifestation of SLE.
- May precede other aspects of SLE by months or even years.
- It has been estimated that 3-15% of patients with apparently isolated ITP go on to develop SLE.
- Chronic and usually asymptomatic
- Acute and severe:
- Occurs as part of disease exacerbation.
- Requires immediate treatment.
- Responds better to glucocorticoids than the chronic and less severe form.
- May occur prior to the development of SLE:
- The only clinical manifestation of thrombocytopenia is bleeding, though severe bleeding from thrombocytopenia is rare.
- SLE and thrombocytopenia associated with:
- Disease activity in general
- Neuropsychiatric manifestations
- Hemolytic anaemia
- Antiphospholipid syndrome
- Renal disease
- aPL, antiribonucleoprotein and anti-Ro antibodies
Treatment
- Most patients with SLE and thrombocytopenia do not require specific treatment.
- in patients with very low platelet counts:
- Glucocorticoid is the first-line therapy
- Danazol
- Hydroxychloroquine
- Intravenous immunoglobulin (IVIG)
- Other immunosuppressants
- vincristine
- Cyclosporine
- cyclophosphamide
- Azathioprine
- biological therapies
- Rituximab
- Splenectomy
- According to the 2019 EULAR guideline recommendations:
- First-line treatment for patients with platelets 30 < ×109/L :
- Moderate/high doses of glucocorticoids (GC) in combination with immunosuppressive agent (AZA, MMF or cyclosporine; the latter having the least potential for myelotoxicity) to facilitate GC-sparing
- Initial therapy with pulses of intravenous methylprednisolone (1–3 days) encouraged.
- Intravenous immunoglobulin (IVIG) may be considered in the acute phase, in cases of inadequate response to high-dose GC or to avoid GC-related infectious complications.
- In patients with no response to GC (ie, failure to reach a platelet count >50,000/mm3) or relapses, RTX should be considered, considering also its efficacy in ITP. Cyclophosphamide may also be considered in such cases. Thrombopoietin agonists or splenectomy should be reserved as last options.
- First-line treatment for patients with platelets 30 < ×109/L :
- The American Society of Hematology 2019 guidelines for immune thrombocytopenia provide recommendations for several types of secondary ITP, but not SLE.
Thrombocytosis
- Defined as platelet count > 450 × 109/L.
- Reactive (secondary) thrombocytosis due to SLE reported in about 4% of patients.1
- Causes include:
- Underlying inflammation
- Iron deficiency anemia
- Autosplenectomy
Red blood cells
Anemia

Definition
- Anemia is defined similarly in patient with and without SLE:
- Hb < 12 g/dL in women
- Hb < 13 g/dL in men
- Anemia not included in the 2019 EULAR/ACR diagnostic criteria.
- 2012 SLICC diagnostic criteria includes hemolytic anemia as a clinical criterion for SLE.
Epidemiology
- Anemia is the most common hematologic abnormality in patients with SLE.
- Present in > 50% of all patients.
- In a cohort of 221 patients with SLE, anemia present in 56.1% of patients at the time of diagnosis.
- In a cohort of 115 patients with SLE, anemia present in 90% of patients.
Pathogenesis
- Causes include:
- Inflammation:
- The most common type of anemia in SLE patients.
- Accounts for approximately a third of cases.
- SLE patients with anemia of inflammation have a significantly higher disease activity than patients with other types of anemia.
- Nutritional deficiency:
- Iron deficiency:
- Present in approximately one-third of patients with anaemia.
- Usually caused by blood loss, especially from gastrointestinal tract.
- Vitamin B12 deficiency:
- Pernicious anemia
- Celiac disease
- Folate deficiency:
- Rare in current era of food fortification.
- Secondary to malnutrition or malabsorption.
- Iron deficiency:
- Medications:
- Cyclophosphamide
- Hydroxychloroquine
- Mycophenolate
- Azathioprine
- Autoimmune hemolytic anemia:
- Reported in 3-8% of patients.
- Included in both the ACR and the SLICC classification criteria.
- May precede SLE diagnosis by several years or coincide with SLE onset.
- A positive DAT is present in 18-65% of SLE patients.
- In a cohort of 628 patients with SLE, 10% of patients developed hemolytic anemia at some time during the disease course, 83% at or before diagnosis. Variables independently associated with degrees of hemolytic anemia were African American ethnicity, thrombocytopenia, and the use of azathioprine.
- Thrombotic microangiopathic hemolytic anemia:
- Thrombotic thrombocytopenic purpura (TTP)
- Drug-induced thrombotic microangiopathy
- Disseminated intravascular coagulation (DIC)
- Catastrophic antiphospholipid syndrome (CAPS)
- Pure red cell aplasia (PRCA):
- SLE is a cause of secondary PRCA; other causes include:
- ABO-incompatible stem cell transplant
- Lymphoproliferative disorders such as chronic lymphocytic leukemia and large granular lymphocyte leukemia (most common)
- Other hematologic conditions such as chronic myelogenous leukemia
- Solid tumors, especially thymoma
- Infections, especially parvovirus B19
- Drugs and toxins
- Pregnancy
- SLE is a cause of secondary PRCA; other causes include:
- Hypersplenism
- Renal insufficiency
- Aplastic anemia
- Hereditary hemolytic anemias such as thalassemia or sickle cell disease
- Inflammation:
- Mechanisms:
- Autoimmune hemolytic anaemia (AIHA) involves anti-red blood cell (RBC) antibodies which damage erythrocytes in either a complement-dependent or independent manner.
- Pure red cell aplasia:
- Autoantibodies directed against developing RBC precursors (or against erythropoietin) interfere with the production of RBCs.
- Microangiopathic hemolytic anemia (MAHA)/thrombotic microangiopathy:
- Anemia from mechanical shearing of RBCs within the circulation, producing schistocytes on the peripheral blood smear.
Clinical presentation
Autoimmune hemolytic anemia (AIHA)
- Approximately two-thirds of cases occur at SLE presentation but AIHA can also present first.
- The presentation of autoimmune hemolysis in SLE is the same as for those without SLE, and includes:
- Anemia with an increased reticulocyte count
- Increased indirect bilirubin and lactate dehydrogenase (LDH)
- Low serum haptoglobin
- Positive direct antiglobulin (Coombs) test
- Spherocytes on the peripheral blood smear
- Associated with:
- Renal disease
- Neurologic disease with seizures
- Serositis
- Presence of aPL
- In some cases, autoimmune hemolysis occurs with ITP (referred to as Evans syndrome):
- Evans syndrome associated with SLE is rare.
- Often precedes the onset of SLE.
Pure red cell aplasia
- Typically diagnosed concomitantly or shortly after SLE diagnosis.
- Patients usually present with:
- Severe normocytic, normochromic anemia
- Very low reticulocyte (reticulocytopenia)
- Aplasia or severe hypoplasia of the red cell line
- White cell and megakaryocyte/platelet lies remain normal
Treatment
Autoimmune hemolytic anemia
- Therapy for autoimmune hemolytic anemia in the setting of SLE is largely based on evidence from individuals without SLE:
- High-dose glucocorticoids – first line therapy
- Rituximab
- Mycophenolate
- Cyclosporine
- Danazol
- splenectomy
- IV immunoglobulin (IVIG)
- According to the British Society for Haematology:
- Treat with corticosteroids as first-line therapy
- Consider immunosuppressive drugs such as azathioprine, mycophenolate mofetil, danazol, or rituximab as second-line therapy
- Splenectomy should only be considered if medical management is ineffective
- According to the 2019 EULAR guideline recommendations: “Autoimmune hemolytic anaemia (AIHA) is far less common than thrombocytopenia in SLE; its treatment follows the same principles regarding use of GC, IS drugs and RTX.”
Pure red cell aplasia
- Management is similar to that of individuals without SLE.
- There is no standard treatment strategy for PRCA.
- Treatment involves:
- Management of the underlying disease
- Red cell transfusions as needed
- First line agents include:
- Corticosteroids
- Cyclosporine A
- Second line agents include:
- Azathioprine
- Cyclophosphamide
- Alemtuzumab
- Antithymocyte globulin
- Rituximab
- Tacrolimus
- IV immunoglobulin (IVIG)
Thrombotic microangiopathic hemolytic anemia:
- Thrombotic thrombocytopenic purpura (TTP) – plasma exchange
- Drug-induced thrombotic microangiopathy (DITMA) – drug discontinuation
- Catastrophic antiphospholipid syndrome (CAPS) – anticoagulation and glucocorticoids
Pancytopenia:
Causes include:
- Hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS)
- Bone marrow toxicity caused by medications or infection
- Hematologic malignancy
- Multiple autoimmune cytopenias
- Splenomegaly with hypersplenism
- Other disorders that occur in the general population, such as vitamin B12 or folate deficiency, myelodysplastic syndrome, and infiltrative tumors
- Autoimmune myelofibrosis:
- Bone marrow findings may be difficult to distinguish from primary myelofibrosis
- Autoimmune myelofibrosis is reported to respond to glucocorticoids.
Lymphadenopathy and splenomegaly
Lymphadenopathy
- Lymphadenopathy (enlargement of one or more lymph nodes) occurs in present in 12%-78% of patients with SLE.
- The nodes are typically:
- Soft
- Nontender
- Discrete
- Varying in size from 0.5 to several centimeters
- Typically in the cervical, axillary, and inguinal areas
- Lymphadenopathy is more frequently noted:
- At disease onset
- In association with a flare
- Biopsies show areas of follicular hyperplasia and necrosis.
- Lymph node enlargement in individuals with SLE can also be due to infection (eg, mononucleosis) or a lymphoproliferative disease such as lymphoma or chronic lymphocytic leukemia (CLL).
- The risks of infections and lymphoproliferative disorders are more common in SLE than in the general population due to immune deficits and dysregulation.
Splenomegaly
- Reported frequency of splenic involvement in SLE varies from 9% to 46%, particularly during active disease.
- In cohort of 3,651 patients (90.2% women, mean age at symptom onset 32 years in women, and 37 years in men) with SLE, presence of splenomegaly in 5.5% of men and in 3.1% of women.
- May result from:
- Polyclonal B-cell activation resulting in increased number of immunoblasts, plasmacytoid lymphocytes and plasma cells.
- Extramedullary hematopoiesis
- Not necessarily associated with cytopenias, although often mild cytopenias may be seen due to splenic pooling of blood and blood cells.
Functional asplenia (autosplenectomy)
- Functional asplenia present in about 18% of patients with SLE patients and thrombocytosis and 0.6% in all SLE patients. 2
- Mechanisms may involve:
- Splenic vasculitis with silent infarction or blockage of the splenic reticuloendothelial system by high levels of circulating immune complexes.
- Multiple thrombotic events within the splenic microvasculature due to the aPL-related coagulopathy.
Lymphoma
Epidemiology
- A number of studies have reported a higher incidence of lymphoma in the SLE population compared with healthy cohort.
- SLE is also associated with an increased risk (4–7 fold) of developing lymphoma when compared to the general population.
- Non-Hodgkin’s lymphoma most common, but risk of Hodgkin’s disease also increased in patients with SLE.
- Relative risk for non-Hodgkin’s lymphoma in patients with SLE ranges from 4.39 to 44.4 in various studies.
- The most common histological type of NHL in SLE is the diffuse large B cell lymphoma (DLBCL) type:
- Constituting 50–65% of the SLE-lymphoma cases.
- Most are of non-GCB subtype.
- Unlike the increased overall cancer risk which is generally confined to Caucasian SLE patients, lymphoma risk in SLE is uniformly higher across all ethnicities.3
Pathogenesis
- Possible mechanisms include:
- Immune dysregulation – constitutive immune activation from prolonged antigen stimulation may result in active lymphocyte proliferation which can eventually terminate into a lymphoma.
- Immunosuppression – most studies have failed to demonstrate a significant risk of lymphoma with use of cyclophosphamide and azathioprine.
- In contrast to RA, persisting disease activity of SLE is not directly associated with the development of lymphoma.
Clinical presentation
- Though NHL in SLE is predominantly nodal, extranodal involvement may also occur.
Treatment
- Treatment typically follows that of lymphoma in patients without SLE, though dose reduction of immunosuppressive agents should be considered.
Hemostasis-thrombosis
Thrombosis
- Patients with SLE have a significantly increased risk of arterial diseases and venous thrombosis.
- Incidence rate:
- General population: 0.7–1.13 per 1000 person-years
- Patients with SLE: 10.5–29 per 1000 patient-years
- Incidence of thrombosis 27 to 43–fold higher than in the general population.
- Thrombosis has been reported in 13.3–22.0% of patients with SLE:
- Most occur within the early years of disease.
- Variations observed between ethnic groups and type of thrombosis.
- Occurs in both those with and without lupus anticoagulant.
- In one study of 219 patients, clinical variables preceding onset of thrombotic event (TE) included:
- Short disease duration
- Traditional risk factors
- Disease activity
- Current and cumulative dose of prednisone
- Lupus anticoagulant (LAC), particularly in combination with anti-RNP/Sm antibodies. Authors conclude: “These findings show that although LAC is involved in the risk of TE, other elements (i.e., inflammation and traditional risk factors) impose the major burden in SLE.”
Antiphospholipid Antibody Syndrome (APS)
Definition
- Systemic autoimmune disorder characterized by thrombotic events or pregnancy morbidity and persistently elevated titers of antiphospholipid antibodies.
- May occur as:
- Primary APS – APS that occurs without any associated condition
- Secondary APS (about 1/3 of cases) – APS associated with another systemic autoimmune disease usually SLE
- Classification based on clinical phenotype:
- Thrombotic APS – characterized by venous, arterial, or microvascular thrombosis.
- Obstetrical APS – characterized by fetal loss after the 10th week of gestation, recurrent early miscarriages, intrauterine growth restriction, or severe preeclampsia.
- Catastrophic APS – rapidly recurring vascular occlusions, predominantly affecting small vessels simultaneously or over a short period of time, and at multiple site.
- Diagnosis based on revised Sapporo classification criteria:
- ≥ 1 of clinical criteria and ≥ 1 of laboratory criteria; clinical and laboratory criteria should be separated by > 12 weeks and < 5 years
- Clinical criteria:
- Vascular thrombosis including:
- Arterial
- Venous
- Small vessel thrombosis in any organ or tissue
- Pregnancy morbidity (1 of):
- ≥ 1 unexplained deaths of morphologically normal fetus ≥ 10 weeks gestational age; normal fetal morphology confirmed by ultrasound or by direct examination of fetus
- ≥ 1 premature births of morphologically normal neonate < 34 weeks gestational age due to eclampsia, severe pre-eclampsia, or placental insufficiency
- ≥ 3 unexplained abortions before 10 weeks gestation (maternal anatomic or hormonal abnormalities and parental chromosomal causes excluded)
- Vascular thrombosis including:
- Laboratory criteria (on ≥ 2 occasions ≥ 12 weeks apart):
- Lupus anticoagulant in plasma
- Immunoglobulin (IgG and/or IgM) anticardiolipin antibody in serum or plasma (titer > 40 glycopeptidolipid [GPL] or monophosphoryl lipid A [MPL] or > 99th percentile) as measured by enzyme-linked immunosorbent assay (ELISA)
- IgG or IgM antibeta-2 glycoprotein 1 antibody in serum or plasma (titer > 99th percentile) as measured by ELISA
Epidemiology
- 40% of patients with SLE have antiphospholipid antibodies (aPLs) – between 50 to 70% of patients with SLE and aPL develop clinical features of antiphospholipid syndrome (APS) after 20 years of follow-up
- About 20% of APS patients reported to have systemic lupus erythematosus (SLE).
Pathogenesis
- aPL can induce a pro-thrombotic status decreasing the thrombophilic threshold by inducing a prothrombotic and proinflammatory phenotype in endothelial cells.
- Risk for thrombosis may be increased by other variables, including:
- Prolonged immobilization
- Pregnancy
- Endothelial dysregulation
- Major target of antiphospholipid antibodies is β2-glycoprotein I (β2GPI), a plasma protein that binds avidly to phospholipid surfaces.
- The binding of antiphospholipid antibodies to β2GPI on cellular surfaces up-regulates the expression of prothrombotic cellular adhesion molecules such as E-selectin and tissue factor, suppresses the activity of the tissue factor pathway inhibitor, reduces activated protein C activity, and activates complement.
Clinical presentation
- Venous thrombosis:
- Deep vein thrombosis (DVT) of the lower extremities is the most frequently reported manifestation.
- Other sites include:
- Superficial veins of legs
- Pelvic veins
- Renal veins
- Hepatic and portal veins
- Inferior vena cava
- Upper extremity veins:
- Axillary
- Subclavian
- Ocular veins
- Cerebral sinuses.
- Arterial thrombosis:
- Cerebrovascular accidents are the most common arterial thrombotic manifestations, usually in the form of a stroke or transient ischemic attack, followed by myocardial infarction.
- Other sites include:
- Peripheral arteries in the legs and arms
- Renal artery
- Aortic arch
- Abdominal arteries
- Among patients with SLE, compared to those without aPL antibodies, those with antibodies have greater prevalence of:4
- Thrombosis
- Pregnancy complications
- Valve disease
- Pulmonary hypertension
- Livedo reticularis
- Thrombocytopenia
- Hemolytic anemia
- Acute or chronic renal vascular lesions
- Moderate or severe cognitive impairment
Treatment
- Treat APS patients with thrombotic manifestations with long-term oral anticoagulation therapy:
- For venous thrombosis:
- Administer vitamin K antagonist (VKA).
- Target INR 2-3.
- Treatment with DOAC not suggested in patients with triple aPL positivity due to high risk of recurrent thrombosis.
- For arterial thrombosis
- Consider VKA over treatment with low-dose aspirin only.
- Treatment with DOAC not suggested in patients with triple aPL positivity due to high risk of recurrent thrombosis.
- For venous thrombosis:
- Treat APS patients with obstetric features with aspirin or combination of aspirin and heparin
Medications
In a cohort of 221 patients with SLE:
- Cumulative cytopenia in 90%:
- Disease-related in 83.4%
- Drug-related in 16.6%
- Azathioprine in 66.7%
- Methotrexate in 15.2%
- Cyclophosphamide in 9.1%
- Mycophenolate mofetil in 9.1%
Hydroxychloroquine
- Recommended for all patients with SLE unless contraindicated.
- Hydroxychloroquine rarely associated with:
- Agranulocytosis
- Anemia
- Aplastic anemia
- Leukopenia
- Thrombocytopenia
- Hydroxychloroquine and other antimalarials including primaquine have been associated with hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. The risk of hemolysis is generally considered low except at high hydroxychloroquine doses in patients with severe G6PD deficiency.
Glucocorticoids
- May lead to:
- Decreased lymphocyte count from direct lymphocytolytic effect
- Neutrophilia
Immunomodulating/immunosuppressive agents:
- Methotrexate (MTX)
- In a randomized trial, 2,391 subjects (adults with known cardiovascular disease and diabetes or metabolic syndrome) randomized to LD-MTX:
- Anemia in 30.2% of patients (often macrocytic) 5
- Leukopenia in 9.2% of patients
- Thrombocytopenia in 8.5% of patients
- Approximately 3% to 4% of patients treated with MTX for rheumatoid arthritis have thrombocytopenia.
- Risk of cytopenias from MTX is greatest in the elderly, those with renal insufficiency, patients with hypoalbuminemia.
- Cytopenias may occur as a late complication, even in patients on a stable dosage, particularly as the result of drug interactions involving MTX .
- MTX may suppress the production of dihydrofolate reductase and decrease tetrahydrofolate levels, resulting in:
- Impaired production of purine nucleotides and thymidylate.
- Reduced for cell replication and DNA synthesis.
- The primary etiology of MTX hematologic toxicity is through the drug’s inhibition of dihydrofolate reductase.
- MTX works mainly in rapidly multiplying cells, such as lymphocytes, which explains its significant anti-inflammatory, immunosuppressive, and apoptosis properties.
- Studies have shown that a potential role for increased platelet apoptosis in mediating MTX-induced thrombocytopenia, resulting in mitochondrial dysfunction.
- DailyMed:
- MTX suppresses hematopoiesis and can cause severe and life-threatening pancytopenia, anemia, leukopenia, neutropenia, and thrombocytopenia.
- Rheumatoid Arthritis Incidence 3% to <10%: Stomatitis, thrombocytopenia (platelet count < 100 x 109/L).
- Incidence 1% to <3%: Rash/pruritus/dermatitis, diarrhea, alopecia, leukopenia (white blood cell count < 3 x 109/L), pancytopenia, dizziness.
- Aplastic anemia, lymphadenopathy.
- In a randomized trial, 2,391 subjects (adults with known cardiovascular disease and diabetes or metabolic syndrome) randomized to LD-MTX:
- Azathioprine:
- Between 2% and 17% of patients develop hematologic toxicity, especially neutropenia
- Leukopenia and thrombocytopenia more common than anemia (may be macrocytic)
- Leukopenia and/or thrombocytopenia are dose-dependent; may occur late in the course of therapy with azathioprine.
- DailyMed (prescribed for rheumatoid arthritis)
- Leukopenia:
- Any degree 28% of patients
- White blood cell count < 2.5 x 109/L in 5.3% of patients
- Leukopenia:
- Mycophenolate mofetil:
- Occasional hematologic toxicities occur, including relatively infrequent leukopenia, anemia, and thrombocytopenia.
- Neutrophil dysplasia characterized by a pseudo-Pelger-Huet anomaly.
- DailyMed:
- Cytopenias, including leukopenia, anemia, thrombocytopenia and pancytopenia are a known risk associated with mycophenolate
- Severe neutropenia (ANC <0.5 x103/mL) developed in up to 2% of kidney transplant patients, up to 2.8% of heart transplant patients and up to 3.6% of liver transplant patients receiving mycophenolate mofetil 3 g daily
- Bone marrow failure, cases of pure red cell aplasia (PRCA) and hypogammaglobulinemia have been reported in patients treated with mycophenolate mofetil in combination with other immunosuppressive agents
- The development of neutropenia may be related to mycophenolate mofetil itself, concomitant medications, viral infections, or a combination of these causes
- Cases of pure red cell aplasia (PRCA) have been reported in patients treated with mycophenolate mofetil in combination with other immunosuppressive agents.
Biologics:
- Belimumab:
- A human immunoglobulin G1λ monoclonal antibody that inhibits the binding of soluble B lymphocyte stimulator to B cells.
- The only biological agent currently approved for the treatment of non-renal SLE.
- Recommended in extrarenal disease with inadequate control (ongoing disease activity or frequent flares) to first-line treatments (typically including combination of HCQ and prednisone with or without IS agents), and inability to taper GC daily dose to acceptable levels.
- Not associated with increased risk of drug-induced cytopenias:6
- Leukopenia: 4.2% (belimumab 10 mg/kg) vs 3.7% (placebo) (pooled analysis of Phase 2, BLISS-52 and 76 studies).
- Neutropenia: 5.2% (belimumab 10 mg/kg) vs 5.5% (placebo) (pooled analysis of Phase 2, BLISS-52 and -76 studies).