Postscript
Introduction
- Acquired hemophilia A is a rare bleeding disorder caused by autoantibodies (also called inhibitors) that deplete or neutralize the activity of endogenous factor VIII (FVIII).
- These autoantibodies interfere with normal hemostasis, leading to bleeding manifestations with varying degrees of severity ranging from asymptomatic to severe, limb-threatening, or life-threatening bleeding.
- Although acquired inhibitors have been reported for all coagulation factors, inhibitors against FVIII are the most common.
- Acquired hemophilia A occurs in both men and women without a previous personal or family history of bleeding.
- Rapid diagnosis and prompt treatment of bleeds are crucial to optimize outcomes.
Classification
- Primary (idiopathic) – about 50% of cases
- Secondary:
- Malignancy (about 7%-18% of cases), including:
- Hematologic
- Solid tumor
- Autoimmune disease (about 10%-17% of cases), including:
- RA
- SLE
- Sjogren syndrome
- Autoimmune thyroid disorders
- Pregnancy – about 1−5% of cases
- Infections
- Vaccinations
- Dermatological diseases
- Drugs:
- Antibiotics
- Immunomodulatory agents:
- Omalizumab
- Alemtuzumab
- Interferon
- Anti seizure medication
- Clopidogrel
- Malignancy (about 7%-18% of cases), including:
Epidemiology
- Incidence of acquired hemophilia A is estimated to be between 1 and 2 cases per million population per year.
- Pregnancy-associated acquired hemophilia A reported in 1 out of 350,000 deliveries in the United Kingdom and 20 cases over 15 years in Italy.
- Two peaks in acquired hemophilia A incidence are observed:
- Younger women (aged 20-40 years) during pregnancy
- Patients > 65 years (its frequency increases with age)
- Median age at presentation is 74-78 years.
- The incidence appears to be similar in males and females (except in the age group from 20 to 40 years because of the pregnancy-associated cases).
Pathophysiology
- Polyclonal autoantibodies (typically IgG1 and IgG4) against FVIII develop in persons with previously normal coagulation factor function through the breakdown of immune tolerance is caused by a combination of genetic and environmental factors.
- Autoantibodies to FVIII appear have been reported to bind to regions A2, A3, and C2 domains of the protein, interfering with its interaction with factor IXa, phospholipids, and von Willebrand factor.
- Kinetics of inactivation of FVIII are second order or exponential, with rapid initial inactivation, followed by slower phase or period of equilibrium in which factor activity can still be measured.
Clinical presentation
- Tends to present as spontaneous bleeding in older males and females or in postpartum women in the absence of a personal or family history of bleeding.
- May present with findings associated with an underlying condition, such as malignancy or autoimmune disease.
- Peripartum cases present at median 3 months postpartum but may occur up to a year following delivery.
- 90% of patients present with spontaneous bleeding:1
- Heterogeneous bleeding phenotype with severity ranging from asymptomatic to severe, limb-threatening, or life-threatening bleeding.
- Bleeding occurs:
- Spontaneously
- After minor trauma
- Following invasive procedures, for example:
- Placement of a venous catheter
- Endoscopic investigations
- Arterial blood sampling
- Intramuscular injections
- Surgical interventions
- Most common sites of bleeding include:
- Subcutaneous/soft tissue (observed in 80% of patients)
- Intramuscular (observed in 45% of patients)
- Retroperitoneal (occurs in 20% of cases)
- Mucosal, including gastrointestinal and genitourinary
- Severe bleeding, including gastrointestinal and intracranial bleeds, can occur.
- Fatal bleeding is usually associated with gastrointestinal bleeding.
- While joint bleeds are a hallmark of congenital hemophilia, they are rare in acquired hemophilia A.
- Compartment syndromes and critical compression of nerves and blood vessels may be seen.
- Bleeding remains a risk until the FVIII inhibitor has been eliminated.
- Acquired hemophilia A may develop in individuals already on anticoagulant therapy.
Diagnosis
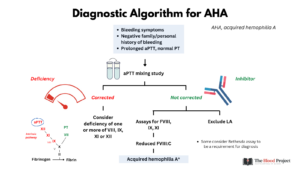
- Check aPTT in a patient with bleeding phenotype.
- An isolated, prolonged aPTT with a normal PT is usually the first indicator of AHA, even in patients without bleeding, and should always be investigated.
- Suspect acquired hemophilia A in patients with unexplained bleeding and a prolonged aPTT.
- Suspicion is further heightened:
- In patients > 60 years of age
- During pregnancy
- Up to on year postpartum
- Screening assays include:
- PT
- aPTT
- Fibrinogen
- CBC
- In a patient with an isolated, prolonged aPTT (with or without bleeding), perform:
- Factor assays:
- FVIII
- One-stage assay
- Chromogenic factor VIII coagulant activity assay
- FIX
- FXI
- FXII
- von Willebrand factor.
- Activity
- Antigen
- FVIII
- Mixing study:
- Used to distinguish a factor deficiency from the presence of an inhibitor.
- Involves performing an aPTT on 50:50 mix of patient’s plasma with normal plasma.
- No correction of aPTT in mixing study suggests the presence of an inhibitor.
- The aPTT is performed/compared at two time points:
- Immediately (immediate mix)
- After 2 hours of incubation (incubated mix):
- In the case of inhibitors to factor VIII, inhibitor activity is time and temperature sensitive and initially overwhelmed by donor factor VIII; inhibitory effects are observed only after some time and at temperatures above ambient room temperature.
- Factor assays:
- Confirm acquired hemophilia A by:2
- The finding of a low FVIII coagulant activity (FVIII:C):
- <1% in 50% of cases
- <5% in 75% of cases
- <40% in 100% of cases
- Plasma mixing study showing failure to correct the prolonged aPTT, especially at 2 hours.
- The finding of a low FVIII coagulant activity (FVIII:C):
- Inhibitor assays:
- If the FVIII level is reduced, a standard Bethesda or Nijmegen-modified Bethesda should be performed.
- Patient plasma is incubated with normal plasma as a source of FVIII and the amount of FVIII measurable in the mixture after 2 hours is compared to a negative control (inhibitor-free FVIII-deficient plasma incubated with normal plasma).
- The dilution closest to a 50% inhibition of FVIII in normal plasma is selected to estimate the inhibitor titer.
- If rpFVIII is an available treatment option, newly diagnosed AHA patients should have another inhibitor assay performed on the same sample, using rpFVIII as the source of FVIII in the Bethesda assay.
- FVIII inhibitor may also be confirmed using an enzyme-linked immunosorbent assay (ELISA) with an anti-FVIII
antibody.

Platton et al, Hematology Am Soc Hematol Educ Program. 2023 Dec 8;2023(1):11-18.
- Italian Association of Hemophilia Centers (AICE) recommendations for diagnosis of AHA and acquired inhibitors of other clotting factors: consider AHA in case of recent-onset, unexpected bleeding in patient without personal and family history of bleeding, who exhibits isolated prolonged aPTT not corrected by addition of normal plasma in 1:1 mixing test following incubation for ≥ 2 hours at 37 degrees C (98.6 degrees F).3
- International recommendations on diagnosis and treatment of acquired hemophilia A: We recommend confirming a diagnosis of AHA by testing FVIII activity and inhibitor concentration using the Bethesda assay and/or an anti-FVIII ELISA.
Differential diagnosis
- A prolonged aPTT may also be caused by:
- Coagulation factor deficiencies
- von Willebrand disease
- Lupus anticoagulant
- Therapy with anticoagulants
- Low FVIII:C may also be caused by:
- von Willebrand disease
- Congenital hemophilia A
- Acquired von Willebrand syndrome
Treatment
- There is little evidence to guide treatment choices. Randomized control trials are not available owing to the rarity of acquired hemophilia A.
- Treatment decisions are often based on the clinical experience of treating clinicians.
- It is recommended that patients with acquired hemophilia A be managed directly by or jointly with specialist hemophilia care, even if patients do not present with severe bleeding.

- Treatment goals include:
- Immediate control of acute bleeding:
- Treatment of acute bleeds is a major priority.
- Not all patients bleed.
- Not all bleeds require intervention; overall, about 70% of patients with acquired hemophilia A need hemostatic treatment.
- There is poor correlation between FVIII:C or inhibitor titer and bleeding phenotype. Thus, hemostatic treatment should be initiated in patients with acquired hemophilia A and clinically relevant bleeding regardless of the patient’s inhibitor titer and residual FVIII activity.
- Indications for hemostatic treatment include:
- Retroperitoneal or retropharyngeal hematomas
- Muscle bleeds
- Intracranial hemorrhage
- Severe hematuria
- Bleeds in multiple sites
- Gastrointestinal bleed
- Pulmonary hemorrhage
- Postoperative bleeding
- Ecchymoses and mild to moderate subcutaneous bleeds may not require hemostatic treatment.
- Appropriate duration of treatment is not established:
- Hemostasis is often achieved within 24 hours to 72 hours.
- Further treatment to prevent recurrence of bleeding may be beneficial.
- Treatment options include:
- First-line treatment (bypassing agent):
- Bypassing agents:
- Options include:
- Recombinant activated factor VII (rFVIIa):
- NovoSeven RT – FDA approved for treatment of bleeding episodes and perioperative management in adults with acquired hemophilia A.
- Administered at an initial dose of 90 µg/kg, with repeat doses administered every 2-3 hours, if necessary, to control bleeding.
- Higher initial dose may be required, for example, 270 µg/kg, depending on the clinical situation but caution needed owing to increased risk of thrombosis.
- aPCC (FEIBA):
- Composed mostly of nonactivated factors II, IX, and X, and activated factor VII (factor VIIa) as well as small amounts of proteins C and S.
- Generally administered at doses of 50-100 U/kg repeated every 6-12 hours, depending on the location and severity of bleeding.
- The maximum recommended dose is 200 U/kg/day.
- Recombinant activated factor VII (rFVIIa):
- Determination of hemostatic control is largely based on clinical assessment.
- After initial hemostasis has been achieved, period of treatment at reduced dose and frequency is often necessary to prevent recurrence and should be evaluated on patient-to-patient basis.
- If initial bypassing agent is ineffective after 12–24 hours, consider switching to the other bypassing agent.
- Treatment with either rFVIIa or aPCC is associated with a risk for arterial and venous thrombosis.4
- Despite the thrombotic risk, treatment should not be withheld since the benefit of early control of severe bleeding outweighs the risk of thromboembolism.
- Recombinant factor VIIa and aPCC should not be administered together except in life- or limb-threatening bleeding unresponsive to individual bypassing agent.
- Options include:
- Bypassing agents:
- Second-line treatment:
- Porcine FVIII:
- Approved for both on-demand hemostatic treatment and prophylaxis in AHA.
- Some patients present with anti-FVIII autoantibodies that cross-react with rpFVIII and reduce efficacy.
- Patients with AHA whose autoantibodies do not cross-react at presentation will often develop a de novo antibody against rpFVIII after ongoing exposure (typically 8 to 85 days after starting rpFVIII).
- 200 U/kg initially with further dosing depending on plasma levels of pFVIII (to maintain trough levels at > 50%) and the clinical situation.
- Factor VIII:
- Human FVIII (hFVIII) replacement, although effective in patients with low titers (<5 BU), is not effective in patients with high titer inhibitors (>5 BU).
- Administer recombinant of plasma-derived human FVIII concentrates only if bypassing agent or recombinant porcine FVIII are unavailable or ineffective and inhibitor titer is low.
- Consider FVIII replacement combined with plasmapheresis and immunoadsorption for severe bleeding or if first-line therapy is unsuccessful.
- Porcine FVIII:
- Emicizumab:
- A recombinant, humanized, bispecific antibody that binds activated factor IX and factor X mimicking the function of FVIII.
- Currently, emicizumab is approved for hemostatic prophylaxis for congenital hemophilia A, but has been used off-label in patients with acquired hemophilia A.
- There is an ongoing prospective multicenter phase 3 study of emicizumab prophylaxis in acquired hemophilia A.5
- Anti-FVIII antibodies do not impact the efficacy of emicizumab.
- Use of activated prothrombin complex concentrates (aPCC) should be avoided in patients receiving emicizumab.6
- Other (adjunct) hemostatic agents:
- Tranexamic acid:
- Consider in all bleeds except urinary tract bleeding (risk for urinary outflow obstruction induced by intraureteral or bladder clot formation).
- However, use with caution if given together with aPCC, especially in older adults and in those with comorbidities associated with high risk of thromboembolism.
- Antifibrinolytic agents may be a useful adjunct to therapy, particularly
for mucosal bleeding, with the exception of renal tract bleeding
- Desmopressin:
- Desmopressin (DDVAP) is of limited utility in AHA.
- Typically reserved for minor bleeding in patients with low inhibitor titers (<2 BU/mL) and FVIII levels >5 IU/L.
- Tranexamic acid:
- First-line treatment (bypassing agent):
- Monitoring treatment:
- Clinical assessment of bleeding.
- There are no specific tests for monitoring treatment with bypassing agents.
- Residual FVIII activity levels and inhibitor titers do not correlate with the severity or responsiveness of bleeds.
- Immediate control of acute bleeding:


Poston J, Kruse-Jarres R. Hematology Am Soc Hematol Educ Program. 2023 Dec 8;2023(1):24-30.
-
- Prevention of of injuries that may provoke bleeding:
- Avoid invasive procedures unless essential owing to the risk of uncontrollable bleeding.
- Surgical interventions and other invasive procedures should be performed only at specialist centers, or with expert advice.
- If a central venous line is required, it may be preferable to use the femoral vein.
- Venipuncture should be performed by experienced staff.
- Blood pressure measurements should be minimized.
- Fasciotomy for intramuscular bleeds should be avoided because this can result in uncontrolled bleeding.
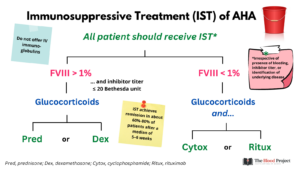
- Long-term eradication of autoantibodies through immunosuppressive therapy (IST):
- Spontaneous remission is rare (about 30% of cases).
- The goal of IST is to reduce the risk of bleeding by shortening the time to achieve remission.
- Owing to the high risk of recurrent bleeding and rarity of spontaneous remissions, immunosuppression is recommended for all patients with AHA irrespective of presence of bleeding, inhibitor titer, or identification of underlying disease.
- Start IST as soon as diagnosis is confirmed.
- First-line therapy:
- Glucocorticoids alone, using either of the following:
- Prednisone 1 mg/kg/day PO for maximum 4-6 weeks followed by tapering
- Dexamethasone 40 mg PO on days 1, 8, 15, 22
- Glucocorticoids combined with one of the following:
- Cyclophosphamide:
- 1.5-2 mg/kg/d PO for a maximum of 6 weeks,
- 1000 mg IV on days 1 and 22
- Rituximab:
- Monoclonal antibody against CD20
- 375 mg/m2 IV weekly for a maximum of 4 cycles
- 100 mg IV on days 1, 8, 15, and 22
- No randomized trials found evaluating efficacy and safety of rituximab for eradication of inhibitors in patients with AHA.
- Recommended as first-line IST in patients with:
- Poor prognostic markers
- In those with contraindications against corticosteroids
- Cyclophosphamide:
- Indications for combined therapy:7
- Patients with FVIII <1%
- Inhibitor titers >20 BU/mL
- Glucocorticoids alone, using either of the following:
- Second-line therapy (if no response in 3-5 weeks):
- Suggested if no reduction in the inhibitor titer or rise in the baseline FVIII level is seen following fter 3-5 weeks of first-line therapy.
- Options include adding cyclophosphamide or rituximab, whichever was not used during first-line therapy, or alternative agents such as mycophenolate mofetil (1 g/day for 1 week followed by 2 g/day), azathioprine, vincristine, cyclosporine.
- IV immunoglobulin (IVIG) as treatment for inhibitor eradication is not recommended.
- Tapering of therapy should be started when inhibitor is undetectable and factor VIII is rising.
- In female patients of reproductive age, avoid cyclophosphamide and other alkylating agents, if possible.
- Monitoring response to IST:
- Inhibitor titer and factor VIII activity levels should be monitored at least weekly during IST until inhibitor becomes undetectable and factor VIII level is normalized.
- IST should be withdrawn or tapered as soon as PR is achieved.
- Prevention of of injuries that may provoke bleeding:
-
- Treat the underlying condition if applicable.
- Transfusions as necessary.
Follow up
- Consider monitoring at least monthly for first 6 months due to common relapses, every 2-3 months up to 1 year, and every 6 months afterwards.
- After remission is achieved continue follow-up for ≥ 12 months because of significant risk of recurrence.
Prognosis
- Partial remission is defined as FVIII >50 IU/dL and no bleeding after stopping any hemostatic treatment for at least
24 hours. - Complete remission is defined as normal FVIII with no detectable inhibitor and either:
- Immunosuppression either stopped or reduced to levels used before AHA diagnosis
- Prednisolone <15 mg/d and all other immunosuppression stopped
- Complete remission is achieved by 60% to 90% of patients.
- Time to achieve remission ranges between a few weeks and many months.
- Approximately 50% of patients experience recurrent bleeding.
- The risk of bleeding is increased as long as the FVIII level remains < 50%.
- Fatal bleeding can occur up to 5 months after first presentation in patients with persistent inhibitors.
- Relapse occurs in approximately 20% of patients, most often early after achieving PR.
- The most common cause of death is infection, secondary to IST
- Other causes of mortality include:
- Cardiovascular disorders
- Underlying disorders
- Bleeding
- Predictors for mortality include:
- Inhibitor titer at baseline
- Advanced age (>75 years)
- Malignancy
- Intensive care unit admission
- WHO performance status
Guideline recommendations



