About the Condition
Description:
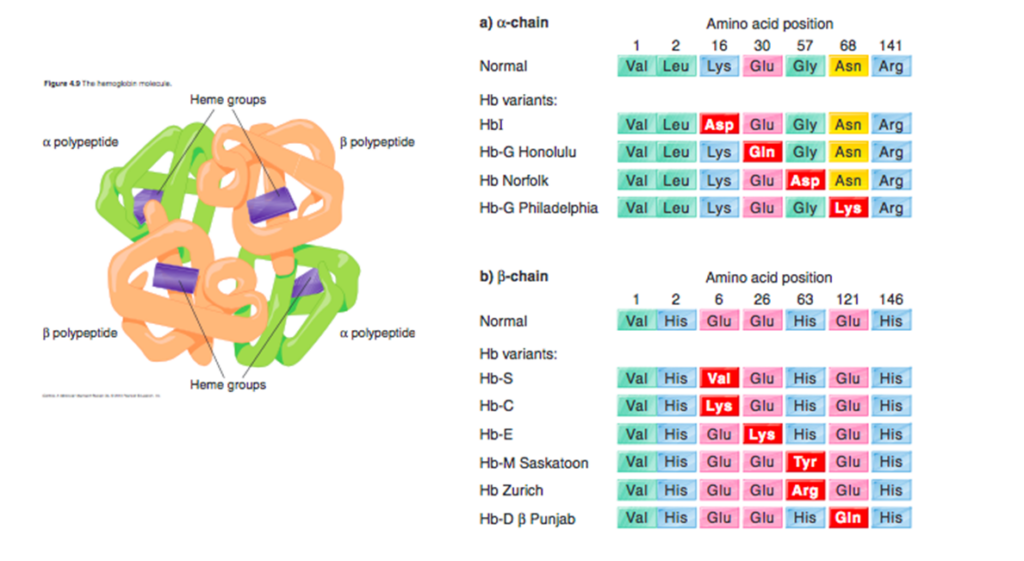
Hemoglobin (Hb) C disorders are a group of chronic, hereditary anemias characterized by the presence of ≥ 1 HbC allele leading to symptomatic disease. HbC is caused by an inherited single base mutation at codon 6 of the beta-globin gene, leading to the substitution of lysine for glutamic acid.
After HbS, HbC is the most common qualitative Hb abnormality in the United States. Frequency of the HbC allele in African Americans in the United States is 1%-2%.
Although HbC disease is mild and does not develop into serious clinical complications, its inheritance with other hemoglobinopathies such as hemoglobin S (HbSC) may have serious consequences.
HbC-related disorders include:
- HbC disease
- Homozygous subtype (HbCC)
- Compound heterozygous subtypes, including:
- HbC/B0-thal
- HbC/B+-thal (like the patient in this case study)
- HbC/Hb Hope
- HbC/Hb Lepore
- HbC/alpha thal
- Sickle cell disease (SCD) – HbSC is a HbC disorder associated with SCD, and accounts for 30% of all SCD.
HbC may protect carriers from dying of malarial infection.

Pathophysiology:
HbC is an inherited single base mutation at codon 6 of the beta-globin gene, leading to substitution of lysine for glutamic acid.
HbCC:
- Reduced solubility of HbC leads to crystal formation rather than hemoglobin polymerization and red cell sickling.
- Associated with high activity of K-Cl transporter, leading to reduced water and potassium content, which results in:
- Increased cell surface: volume ratio, resulting in target cells.
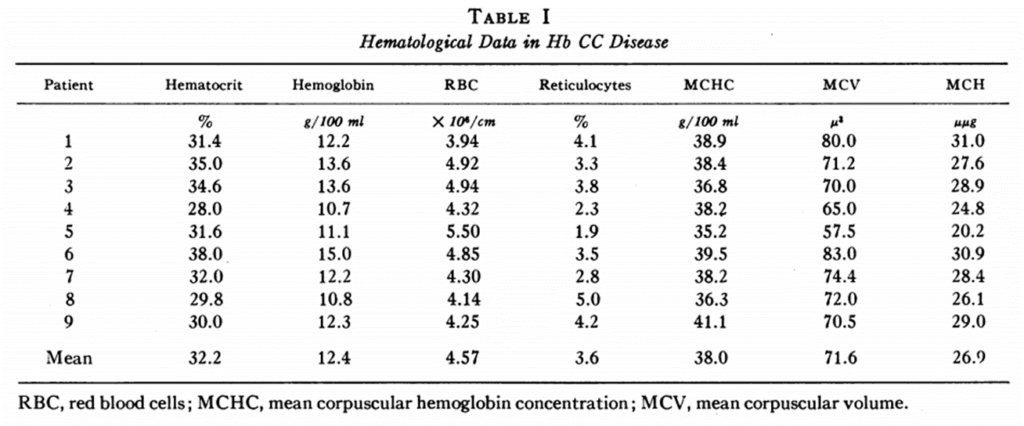
- Dehydrated cells, which are denser than normal, microcytic, and hyperchromic with increased mean corpuscular hemoglobin concentration (MCHC).
- Crystallization and cell dehydration lead to hemolysis and hyperviscosity, but not vaso-occlusion.
- Red cell life span is shortened to approximately 40 days in HbC disease. This is at least three times as long as the lifespan of cells in sickle cell anemia.
HbSC:
- HbC-mediated dehydration of erythrocytes leads to polymerization of HbS and red cell sickling.
- Higher hematocrit in HbSC vs. HbSS leads to increased viscosity, which may contribute to increased incidence of certain complications such as retinopathy.
Clinical presentation:
Hemoglobin C trait (HbAC) is clinically silent.
Patients homozygous for HbC (HbCC, hemoglobin C disease) may present with:
- Mild hemolytic anemia
- Splenomegaly
Patients who co-inherit HbC and HbS (HbSC) have a sickle cell disease phenotype including:
- Acute chest syndrome
- Vaso-occlusive crisis
- Splenic sequestration
- Stroke
- Priapism
- Retinopathy
- Avascular necrosis
Diagnosis:
HbCC and HbSC typically diagnosed in childhood (newborn screening is mandated in the United States).
Suspect diagnosis of Hb CC in individual with:
- Complete blood count showing:
- Microcytosis
- Elevated mean corpuscular hemoglobin concentration – average MCHC in HbC disease, HbSC disease, HbAC trait, and HbA cells 38, 37, 34, and 33 g/dl, respectively.
- Peripheral smear showing:
- Target cells
- Microspherocytes
- Polychromatophilic cells
- Hemoglobin crystals (crystalline inclusions)
- Hyperchromic red cells
- Physical exam, labs and/or imaging showing:
- Mild hemolytic anemia
- Splenomegaly
- Jaundice (infrequent)
Suspect diagnosis of Hb SC in individual with:
- Presentation similar to but less severe than hemoglobin SS disease, especially vascular retinopathy and avascular necrosis of the femoral head.
- Normal of near normal Hb.
- Peripheral smear showing:
- Target cells
- Hemoglobin crystals (may be absent if concomitant alpha-thalassemia)
- Irreversibly sickled cells (rare)
Confirm diagnosis of HbCC and HbSC by:
- Isoelectric focusing (IEF)
- Hemoglobin electrophoresis (HbEp)
- High performance liquid chromatography (HPLC)
- DNA analysis
Treatment:
In patients with HbCC, treatment not typically required.
In patients with HbSC, treatment recommendations are generally the same as in sickle cell anemia.