About the Condition
Description/definition:
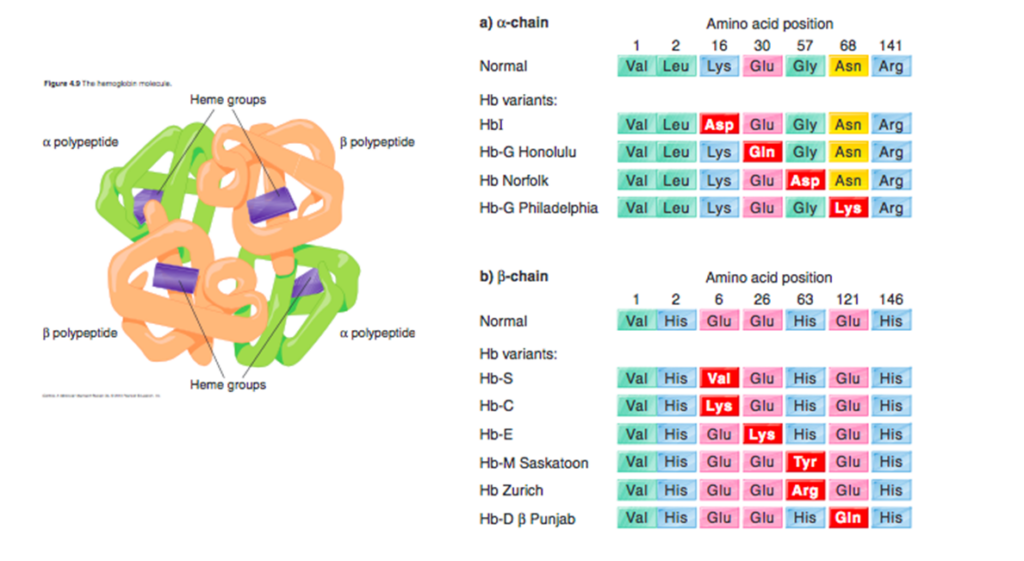
HbE disorders are a group of chronic, hereditary anemias characterized by presence of ≥ 1 hemoglobin E (HbE) allele, often leading to symptomatic disease. HbE is an inherited single base mutation at codon 26 of the beta-globin gene, leading to substitution of lysine for glutamic acid. Because synthesis of the beta chain of HbE is impaired, HbE is considered a type of beta+-thalassemic hemoglobinopathy.
Individuals with HbE trait (HbAE) and HbEE (also called HbE disease) are asymptomatic, although they may have very mild anemia and microcytosis.
Patients with HbE and co-inherited beta-thalassemia have a wide spectrum of disease ranging from nontransfusion-dependent beta-thalassemia to transfusion-dependent beta-thalassemia.
HbE occurs at an extremely high frequency in many countries in Asia. The frequency of Hb E approaches 60% in some regions of Thailand, Laos and Cambodia.
HbE β-thalassemia has replaced β-thalassemia as the most common thalassemia disorder in many of these regions.

Pathophysiology:
HbE is an inherited single base mutation at codon 26 of the beta-globin gene (GAG-AAG), leading to substitution of lysine for glutamic acid.
The mutation activates a cryptic mRNA splice site, which results in reduced synthesis of the β-E chain and leads to a mild globin-chain imbalance, similar to that observed in beta thalassemia heterozygotes. Thus, it is considered to be a thalassemia phenotype.
In addition, Hb E has a weakened α/β interface, leading to some instability during conditions of increased oxidant stress.
HbE is found in several compound heterozygotes:
- Beta-thalassemia:
- HbE beta+-thalassemia
- Hb E-β0-thalassemia
- Hb E δβ-thalassemia
- Hb E Lepore
- Hb SE
- HbE Constant Spring
- HbEF Bart
- HbAE Bart
In HbE beta-thalassemia, globin chain imbalance results in precipitation of alpha globin chains and ineffective erythropoiesis, apoptosis, oxidative damage, and shortened red cell survival.
The resistance of Hb AE red cells to invasion by Plasmodium falciparum is most likely the cause for its high prevalence throughout the world.
Clinical presentation:
HbE alone does not causes clinical disease; clinical problems arise when HbE is co-inherited with various forms of beta-thalassemia or alpha-thalassemia:
- Individuals with HbAE trait are asymptomatic. They may have mild microcytosis without anemia. Mean corpuscular hemoglobin (MCH) is low, mean corpuscular hemoglobin concentration (MCHC) is normal.
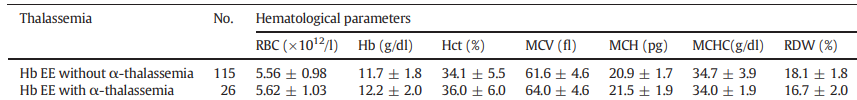
- HbEE individuals are asymptomatic with occasional splenomegaly and very mild anemia with microcytosis. MCH is low, MCHC is normal. The red cell morphology may show targeting.
- Patients with HbE and co-inherited beta-thalassemia have a wide spectrum of disease ranging from nontransfusion-dependent beta-thalassemia to transfusion-dependent beta-thalassemia. They may present with hepatosplenomegaly, extramedullary hematopoietic masses, growth retardation, delayed sexual maturation, and bone deformities. Iron overload in nontransfused patients is common.
| HbE | Hb | MCV | Clinical |
|---|---|---|---|
| Hb E heterozygote | 12.8 + 1.5 | 84 + 5 | Normal |
| Hb E homozygote | 10.6 + 1.2 | 65 + 3 | Normal |
| Hb E b thalassemia | 7.1 + 1.4 | 59 + 3 | Mild-severe disease |
Diagnosis:
Consider diagnosis of:
- HbAE and HbEE if:
- Complete blood count (CBC) shows mild anemia and microcytosis.
- Peripheral smear reveals target cells and irregularly contracted cells.
- HbE beta-thalassemia if:
- CBC shows anemia and microcytosis.
- Steady state hemoglobin varies from 3 g/dL (30 g/L) to 11 g/dL (110 g/L).
- Peripheral smear may reveal target cells and irregularly contracted cells.
Confirm diagnosis using qualitative and quantitative analysis of hemoglobin:
- HbAE:
- About 30% HbE and 70% HbA.
- HbE is less than 50% because it is synthesized at a slightly reduced rate and because alpha-globin chains have lower affinity for betaE than betaA chains.
- HbEE:
- HbE ≥ 90%.
- Mild elevation in HbF (0%-10%).
- HbE beta-thalassemia:
- HbE about 40%-60% with significant elevation in HbF in HbE beta0-thalassemia.
- HbE and variable amounts of HbA (typically about 10%) in HbE beta+-thalassemia.
Qualitative and quantitative analysis of hemoglobin:
- Hemoglobin (Hb) electrophoresis:
- In alkaline Hb electrophoresis, HbE migrates with Hb C, O Arab, and A2.
- In acid pH electrophoresis, HbE migrates with HbA2.
- Isoelectric focusing, HbE migrates close to Hb O Arab.
- High-performance liquid chromatography (HLPC), HbE has similar retention times as HbA2 and Hb Lepore.
DNA testing
Treatment:
Patients with HbAE and HbEE are asymptomatic and do not require treatment.
Patients with HbE beta-thalassemia may require:
- Red cell transfusions
- Iron chelation therapy
- Treatment of organ damage from iron overload (especially cardiac, endocrine, pulmonary, hepatobiliary, and skeletal systems)
- Treatment of other complications including:
- Infections
- Thromboembolism
- Jaundice, gallstones, and cholecystitis