Labs
The following is the patient’s complete blood count (CBC) at the time of admission:
| WBC (109/L) | Hb (g/dL) | MCV (fL) | PLT (109/L) |
|---|---|---|---|
| 6.8 | 13.6 | 87 | 398 |
What’s what: WBC, white blood cell count; Hb, hemoglobin; MCV, mean cell volume; MCHC, mean cellular hemoglobin concentration; RDW-SD, red cell distribution width-standard deviation; platelets, PLT; Normal values: WBC 5-10 x 109/L, RBC 4-6 x 1012/L, Hb 12-16 g/dL, Hct 35-47%, MCV 80-100 fL, MCHC 32-36 g/dL, RDW-SD < 45 fL, platelets (PLT) 150-450 x 109/L
Let’s look at how a PT and aPTT are carried out:
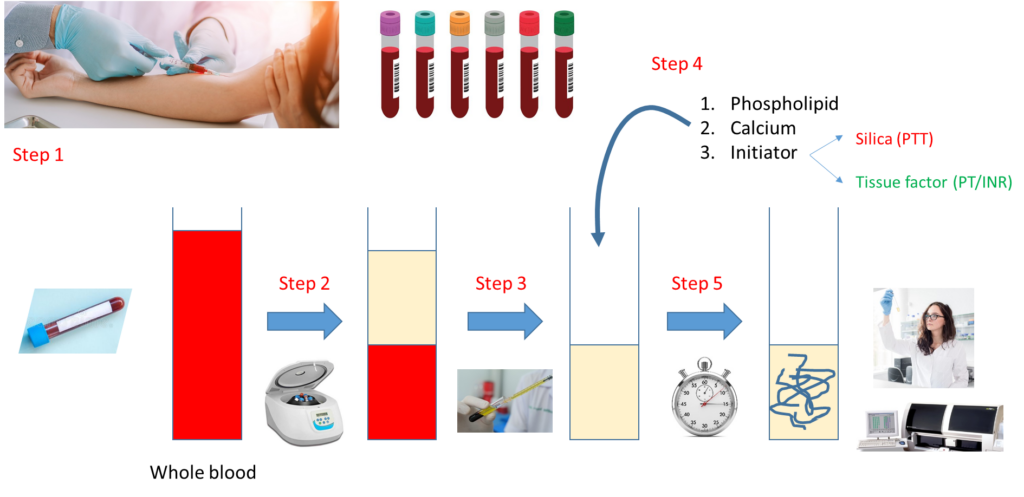
Notes:
- Step 1 – Draw whole blood from a blood vessel (typically an arm vein) into a blue top tube containing the anticoagulant, sodium citrate.
- Step 2 – Spin the liquid blood sample in a centrifuge so that red cells layer on the bottom, plasma on the top.
- Step 3 – Pipette plasma (top layer) into a clean, empty test tube.
- Step 4 – Add phospholipid, calcium and activator (tissue factor for PT, silica for aPTT).
- Step 5 – Incubate at 37 degrees C and measure time to clot formation, either with automated instrument or more rarely by vision.
Note that the major difference between the PT and aPTT is the nature of the activator added to the sample.
Further work-up of this case requires some knowledge of the clotting cascade. Let’s review the basics:
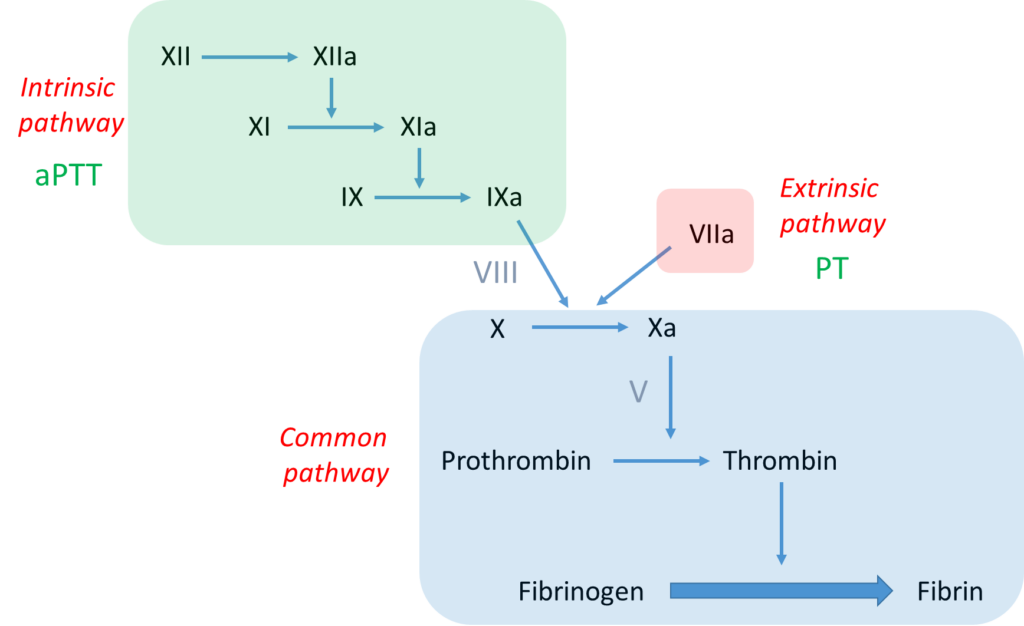
Notes:
- The bottom line is formation of an insoluble fibrin plug, which, along with platelets, stems blood loss.
- Fibrin is derived from thrombin-mediated cleavage of soluble fibrinogen (which is a structural protein).
- Thrombin (which is a type of enzyme called a serine protease) is formed when another serine protease, activated factor X (FXa), cleaves prothrombin.
- Two pathways may activate factor X:
- The intrinsic pathway, which consists of a series of linked reactions involving serine proteases FXII, FXI and FIX, and cofactor FVIII.
- The extrinsic pathway, which consists of tissue factor-mediated activation of FVII (FVIIa).
- In vivo, the clotting cascade is always initiated by tissue factor activation of FVII (extrinsic pathway) and amplified by the intrinsic pathway via cross-talk (FVIIa activates FIX) and feedback (thrombin activates FXI and FVIII) mechanisms.
- aPTT measures the integrity of the intrinsic pathway.
- PT/INR measures the integrity of the extrinsic pathway.

According to the scheme above, an isolated elevation in the aPTT indicates a deficiency of or inhibitor against a clotting factor in the intrinsic pathway, namely FXII, FXI, FIX or FVIII.
Let’s review the basics of the mixing study:

Notes:
- Mixing study involves mixing patient plasma and pooled normal plasma in 1:1 ratio.
- The resulting mixture is then processed for clotting time (PT or aPTT) immediately (time 0) and then after 2 hours of incubation.
- A clotting factor must drop below about 30% before the PT or PTT begins to prolong.
- Therefore, even if a clotting factor is completely missing (i.e. 0% level), the 1:1 mix will restore it to 50% (equal volume of plasma containing 0% of the factor and normal pooled plasma containing 100% of the factor), more than enough to restore a normal PT or aPTT (the example above shows 10% activity of imaginary factor Y in patient plasma, with 1:1 mix resulting in 60% activity).
- However, if there is an inhibitor or antibody to a clotting factor, the inhibitor will carry over from patient to normal plasma in the mix and interfere with factor activity, leading to continued prolongation of the PT or aPTT.
To summarize at this point, we have a patient with excessive postoperative bleeding and a prolonged aPTT that corrects with normal plasma (a negative inhibitor screen or normal mixing study). These results indicate deficiency of one or more clotting factors rather than an inhibitor against a clotting factor.
AVWS, acquired von Willebrand syndrome
Results of the von Willebrand (vWF) screen:
vWF antigen 29% (normal 60-158%)
vWF activity < 12.5% (normal 50-200%)
About von Willebrand factor (vWF) screening tests:
The antigen test is an immunoassay that measures the concentration of vWF protein in plasma.
The activity assay is also called von Willebrand factor ristocetin cofactor (vWF:RCo) activity. It is a functional assay that measures the ability of ristocetin to promote platelet aggregation in the presence of vWF.
Laboratory findings in acquired von Willebrand syndrome (AVWS) are similar to those in von Willebrand disease (VWD) and may include decreased values for VWF:Ag, VWF:RCo, or FVIII. The VWF multimer distribution, which can be ordered as a separate test often shows a decrease in large multimers similar to the pattern seen in type 2A VWD.
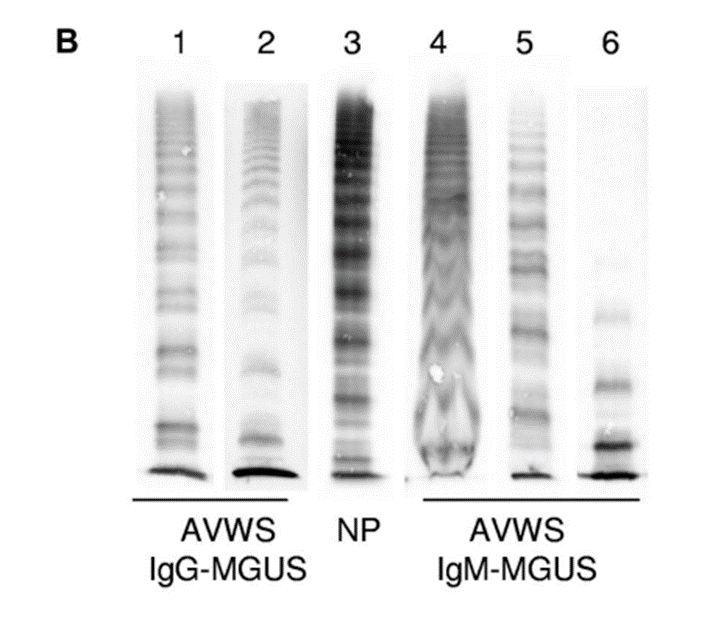
In this case, the vWF antigen level was low, and the vWF activity even lower. In other words, the vWF:RCo/vWF:Ag ratio is reduced. A low vWF:RCo/vWF:Ag ratio is seen in hereditary type 2A VWD where there is a reduction in high molecular weigh VWF multimers. It is also low in many patients with acquired VWS, because they are too are deficient in high molecular weight vWF multimers.

This patient had excessive bleeding around the time of his hernia repair. However, that has resolved without requiring any hemostatic treatment. He is stable and ready to be discharged home.
This patient had an SPEP. The result was reviewed by the hematologist on a follow-up clinic visit:

In summary, the patient has a monoclonal IgM gammopathy as defined by serum protein electrophoresis (SPEP) in combination with immune fixation. The differential diagnosis of monoclonal IgM gammopathy includes IgM-MGUS (monoclonal gammopathy of unclear significance), Waldenström’s macroglobulinemia (WM), and IgM myeloma.
- IgM MGUS is defined by an IgM serum protein of less than 3 g/dL, less than 10% clonal lymphoplasmacytic cells in the bone marrow, and the absence of symptoms typical of WM.
- WM is defined by a monoclonal IgM serum protein and at least 10% monoclonal lymphoplasmacytic cells in the bone marrow clot section or biopsy.