Labs
The following is the original complete blood count (CBC) on blood at room temperature (1) and warmed blood (2):

Let’s reorganize the CBC on the warmed blood sample according to our trusted TBP format:
| WBC (109/L) | Hb (g/dL) | Hct (%) | MCV (fL) | RDW-SD (fL) | PLT (109/L) |
|---|---|---|---|---|---|
| 5.3 | 8.0 | 27.2 | 78 | 52 | 234 |
What’s what: WBC, white blood cell count; Hb, hemoglobin; MCV, mean cell volume; MCHC, mean cellular hemoglobin concentration; RDW-SD, red cell distribution width-standard deviation; platelets, PLT; Normal values: WBC 5-10 x 109/L, RBC 4-6 x 1012/L, Hb 12-16 g/dL, Hct 35-47%, MCV 80-100 fL, MCHC 32-36 g/dL, RDW-SD < 45 fL, platelets (PLT) 150-450 x 109/L
A question for the more experienced:
For more information on how cold agglutinins interfere with the CBC, click here.
What is the first lab test (other than peripheral smear) we should obtain in a patient with normocytic anemia?
Click for Answer
So, the patient appears on the “appropriate reticulocyte count” side of the diagnostic ledger:
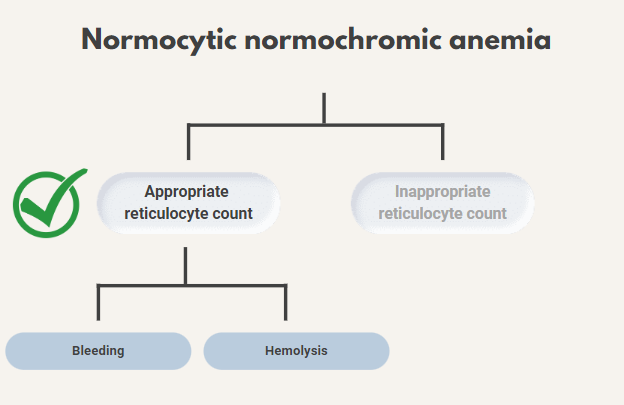
He shows no signs of bleeding, so you decide to focus on the possibility of hemolytic anemia.

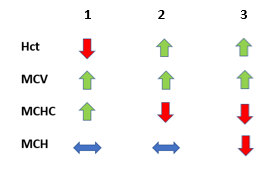
Sort the lab parameters according to their expected blood levels in hemolysis?
Thus far, the data support a diagnosis of hemolytic anemia. Before we consider the peripheral smear, let’s discuss an approach to hemolysis.
What is hemolytic anemia?
In normal physiology, red cells last approximately 120 days in circulation. As they circulate, they gradually develop wear-and-tear damage as they pass through small capillaries or across cardiac valves, as well as oxidative damage to their membranes. Eventually, this damage makes them less flexible. They become unable to navigate the narrow splenic sinuses, and are eventually phagocytosed by splenic macrophages.
“Hemolysis” is an umbrella term that refers to the premature destruction of red blood cells – they are destroyed before reaching the point of physiologic, senescent death. There are a wide variety of processes that can lead to hemolysis, which we will explore below.
What can cause hemolysis?
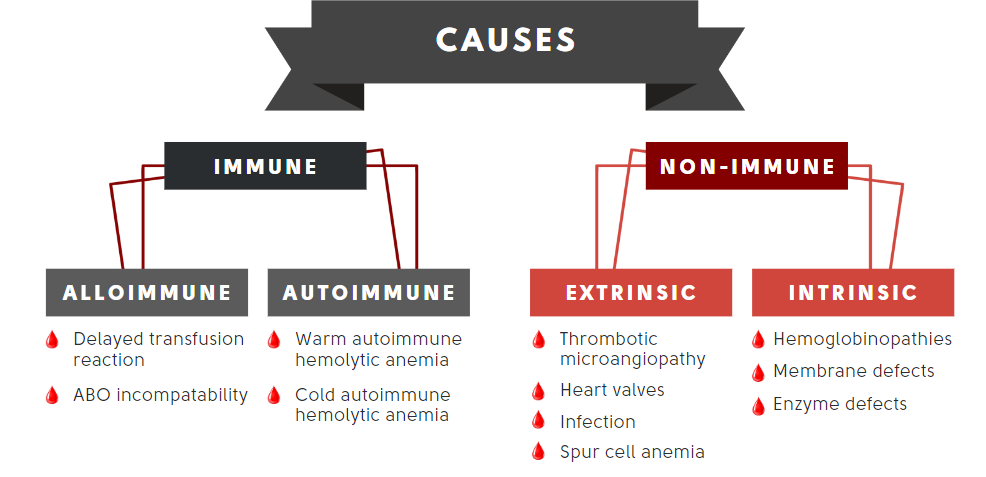
Key point: there are a wide variety of causes. It helps to have a systematic approach.
2. A diagnostic schema offers a systematic approach to generating a differential diagnosis for a particular problem. Schemas generally subdivide your differential into various physiologic “buckets”. We will explore a diagnostic schema for hemolysis on the next slide.

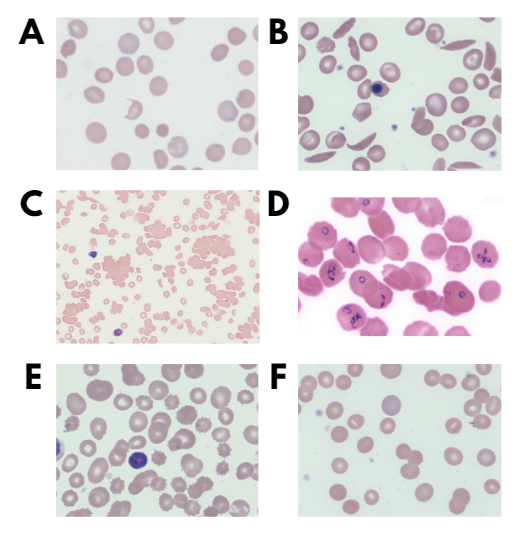
Match the smear (A-C)) with the condition:
Match the smear (D-F) with the condition:
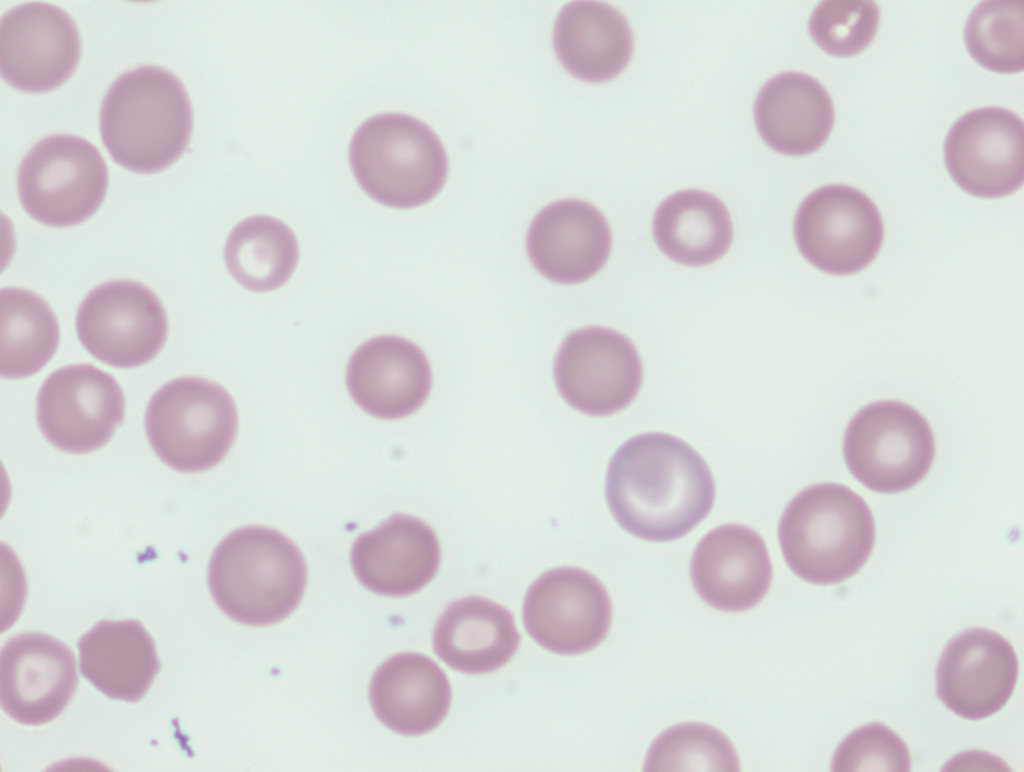
The patient’s smear performed on warmed blood is similar to the following (want to show smear with clumping at room temperature?):
Sort the cause of hemolysis with the predominant mechanism:
The ability of hemolytic labs to differentiate between intravascular or extravascular hemolysis is by no means perfect. At first glance, we might assume that intravascular causes of hemolysis lead to elevated leakage products including LDH, AST and plasma hemoglobin (released into the circulation by lysed cells), while extravascular causes of hemolysis lead primarily to increased indirect bilirubin (from intracellular conversion of Hb to bilirubin).
However:
- Many cause of hemolysis are associated with both intravascular or extravascular hemolysis
- Both intravascular or extravascular hemolysis may be associated with a similar biochemical profile:
- Intravascular hemolysis causes release of:
- LDH
- AST
- Hemoglobin (Hb)
- Free Hb binds haptoglobin (Hp) leading to reduced levels of Hp.
- Hb-Hp complexes are taken up by macrophages, converted to indirect bilirubin.
- Extravascular hemolysis leads to:
- Macrophage engulfment of red blood cells.
- Intracellular degradation of red cell Hb into bilirubin.
- Leakage of LDH and AST back to the circulation.
- Intravascular hemolysis causes release of:


There are two types of cold agglutinin disease:
- Primary (idiopathic) cold agglutinin disease (CAD):
- > 90% of patients reported to have clonal expansion of kappa-positive B cells in bone marrow and a monoclonal IgM-kappa paraprotein.
- Monoclonal cold autoantibody is often encoded by IGHV4-34 and targets I antigen.
- Secondary CAD (referred to by some as cold agglutinin syndrome):
- Arises in the setting of an underlying disorder such as
- Viral infection
- Autoimmune disorder
- Systemic lupus erythematosus (SLE)
- Rheumatoid arthritis
- Overt lymphoid malignancy
- Antibody is typically IgM and its target depends on underlying condition:
- Mycoplasma pneumonia – polyclonal IgM against I antigen
- Epstein-Barr virus – polyclonal IgM against i antigen
- Cytomegalovirus – polyclonal IgM against i antigen
- Aggressive non-Hodgkin lymphoma (NHL) – monoclonal IgM against I antigen, but light chain restriction can be lambda as well as kappa
- Arises in the setting of an underlying disorder such as
Younger patients may be more likely to have an underlying infection or autoimmune disorder, and older individuals (eg, >60 years of age) may be more likely to have a lymphoid (B-cell or plasma cell) malignancy such as aggressive non-Hodgkin lymphoma or Waldenström macroglobulinemia (WM).
Of note, infectious and inflammatory causes tend to result in a temporary surge in polyclonal antibody production, and therefore tends to produce a less severe clinical picture.
In contrast, lymphoproliferative disorders tend to result in monoclonal anti-I production, which can result in a more significant degree of anemia and symptom burden.
