About the Condition
Description/definition:
Cold autoimmune hemolytic anemia (AIHA) is a condition of acquired uncompensated destruction of red blood cells (RBCs) by autoantibodies to RBC antigens that bind optimally to RBCs at 0-4 degrees C (32-39.2 degrees F), but also react at temperatures above 30 degrees C (86 degrees F).
Cold autoimmune hemolytic anemia (AIHA) includes:
- Cold agglutinin disease (CAD)
- Paroxysmal cold hemoglobinuria
CAD is traditionally classified as:
- Primary or idiopathic
- > 90% of patients with primary CAD are reported to have clonal expansion of kappa-positive B cells in the bone marrow and a monoclonal immunoglobulin M (IgM)-kappa paraprotein
- Secondary or acquired, caused by
- Infection
- Malignancy, especially B cell lymphoproliferative disorders
- Autoimmune disease
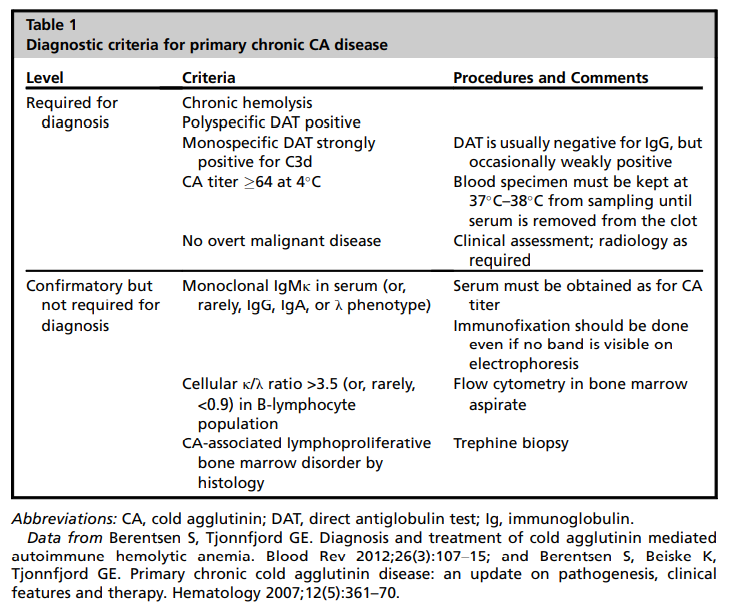
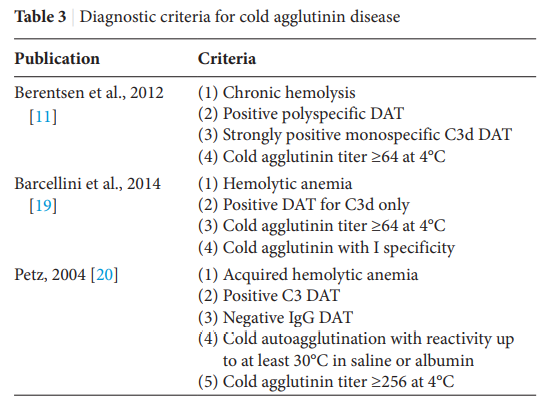
According to a recent international consensus, CAD is defined as “an AIHA with a monospecific direct antiglobulin test (DAT) strongly positive for C3d (and negative or weakly positive with IgG [immunoglobulin G]) and a cold agglutinin (CA) titer of 64 or greater at 4°C. We recognize that there may be occasional cases with CA titer<64. Patients may have a B-cell clonal lymphoproliferative disorder (LPD) detectable in blood or marrow but no
clinical or radiological evidence of malignancy.”1
CAD accounts for approximately 15–25% of autoimmune hemolytic anemias (AIHAs). Prevalence of 5 to 20 cases per million and an incidence of 0.5 to 1.9 cases per million per year.

Pathophysiology:
Cold agglutinin disease caused by complement-fixing antibody (monoclonal immunoglobulin M [IgM] in about 90% of cases) that typically binds to antigens of the I blood group system (rarely the Pr antigen) with maximal reactivity between 0 and 4 degrees C (32-39.2 degrees F), leading to complement-mediated extravascular (and to lesser extent intravascular) hemolysis. Approximately 90% of cold agglutinins have anti-I specificity, while the remaining usually have anti-i specificity.
In primary cold agglutinin disease (CAD):
- Bone marrow displays a characteristic histomorphologic, immune phenotypic, and molecular pattern that has been designated CA-associated LPD.
- > 90% of patients reported to have clonal expansion of kappa-positive B cells in bone marrow and a monoclonal IgM-kappa paraprotein
- Monoclonal cold autoantibody is often encoded by IGHV4-34 and targets I antigen
Clinical Presentation:
- Onset is usually gradual
- Patients may present with symptoms and signs of anemia, including
- Fatigue (attribute to both anemia and complement activation)
- Weakness
- Dizziness
- Dyspnea
- Pallor
- Patients may present with symptoms and signs of hemolysis, including
- Jaundice
- Dark urine
- Hepatosplenomegaly
- Splenic fullness
- Patients may also present with history of cold-induced symptoms, resulting from agglutination of erythrocytes in the acral circulation, including:
- Episodic acute hemolysis; including dark colored urine after exposure to cold, febrile illness, or major trauma
- Using warm clothing or staying indoors in cold weather
- Seasonal variation in symptom severity in cool climates
- Acrocyanosis – in 40-50% of patients
- Raynaud’s phenomenon
- Livedo reticularis
- Patients may present with additional symptoms of underlying disease
- Complement-driven exacerbation of hemolysis is common during febrile infections and other conditions with acute-phase reaction.
- Physical findings include:
- Pallor
- Jaundice
- Splenomegaly (rare)
- Acrocyanosis (bluish appearance of extremities such as toes, fingers, tip of nose, or ears)
- For further information about the physical diagnosis in cold agglutinin disease, click here.
- Labs show:
- Anemia
- About 25% found to have severe anemia, defined as hemoglobin (Hb) < 8.0 g/dL.
- About 35% have moderate anemia, defined as Hb 8.0-10.0 g/dL).
- About 25% have mild anemia (Hb > 10.0 g/dL).
- About 10% have compensated hemolysis (no anemia).
- Hemolysis
- Blood tests
- Urine tests – hemoglobinuria has been reported in at least 15% of the patients.
- Anemia
- Positive DAT (see next section)
Diagnosis:
Clinicopathological diagnosis based on demonstrating hemolytic anemia and serological evidence of ant-red blood cell autoantibodies by direct antiglobulin test (DAT).
Suspect diagnosis in patient with:
- Compatible history
- Unexplained chronic anemia (median hemoglobin of 9-10 g/dL in the US population)
- Chronic hemolysis
Confirm diagnosis if:
- DAT positive for complement with or without immunoglobulin (Ig) G – up to 22% of CAD cases may have a positive anti-IgG DAT, in addition to a positive anti-C3d DAT.
- High titer cold agglutinin
- >1:64 or more.
- Semi-quantitative assay based on ability of antibodies to agglutinate erythrocytes at 4°C).
- Note: most defining criteria of CAD do not require that anti-I/anti-i specificity be present.
Optional confirmatory tests:
- SPEP showing monoclonal IgM-kappa in serum.
- Cellular kappa to lambda ratio > 3.5 in B cells (fluorescence activated cell sorting [FACS]).
- Cold agglutinin-associated lymphoproliferative disorders on bone marrow histology.
Once diagnosis of CAD is established, evaluate for secondary conditions; tests may include:
- Serum Ig and electrophoresis with immunofixation to identify underlying clonal bone marrow lymphoproliferative disorder often associated with primary CAD.
- HIV, hepatitis B, and hepatitis C testing.
- Anti-double-stranded DNA and antinuclear antibody (ANA) testing to help diagnose autoimmune disorders such as systemic lupus erythematosus (SLE).
- Computed tomography (CT) scan of chest, abdomen, and pelvis to rule out malignant disease.

Treatment:
- Treat if symptomatic anemia, severe circulatory symptoms, transfusion dependence.
- > 70% of patients reported to require therapy.
- Treatment decisions are based on expert opinion; prospective studies to support benefit are lacking.
- Nonpharmacological measures (especially avoidance of cold) are cornerstone of management. Has been shown to alleviate symptoms and may prevent severe exacerbations of hemolytic anemia.
- Supportive transfusions may be required
- Consider using blood warmer when transfusing RBCs.
- Blood bank testing for underlying alloantibodies and crossmatch testing should be performed at 37°C.
- If the cold agglutinin has a broad thermal amplitude, obtaining crossmatch compatible blood may be a clinical challenge.
- Therapies may be aimed to suppress production of aberrant IgM or to inhibit complement-mediated destruction of red blood cells (RBCs)
- Consider corticosteroids (prednisolone 1 mg/kg/day) or plasmapheresis as a temporary measure to remove IgM autoantibodies, if anemia is severe or life-threatening.
- Although corticosteroids are a rapid and effective treatment for warm AIHAs, they have not been shown to be as effective in CAD. Retrospective studies have found that <15% of patients respond to corticosteroids, and they require higher doses to maintain remission.
- Typically treated with one of the following regiments:
- Rituximab
- Rituximab plus bendamustine
- Rituximab plus fludarabine
- Consider complement system-targeting monoclonal antibody:
- Eculizumab
- Sutimlimab-jome (Enjaymo), which has been FDA approved to decrease need for RBC transfusion due to hemolysis in adults with CAD.
- Surgical intervention, including splenectomy, has been shown to be effective only in patients with IgG-mediated CAD, which represents only approximately 3.5% of patients.
Prognosis:
- The median overall survival of patients with CAD has been estimated to be 12.5 years, similar to that of a general age- and sex-matched population.
- Estimated risk of transformation of the lymphoproliferative bone marrow disorder to aggressive lymphoma is about 3% to 4%.
- Increased risk of venous thromboembolism.
Tables related to classification and diagnosis