Preparing to see the patient
Hemolytic markers directly released from red blood cells:
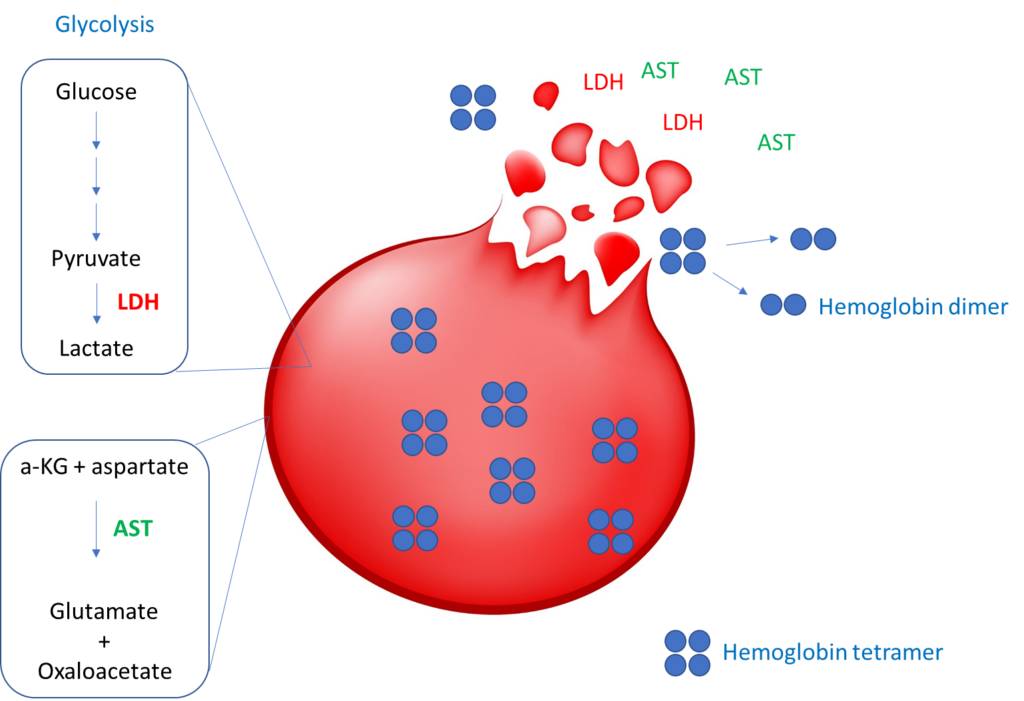
Indirect hemolytic markers (not released by red cells):
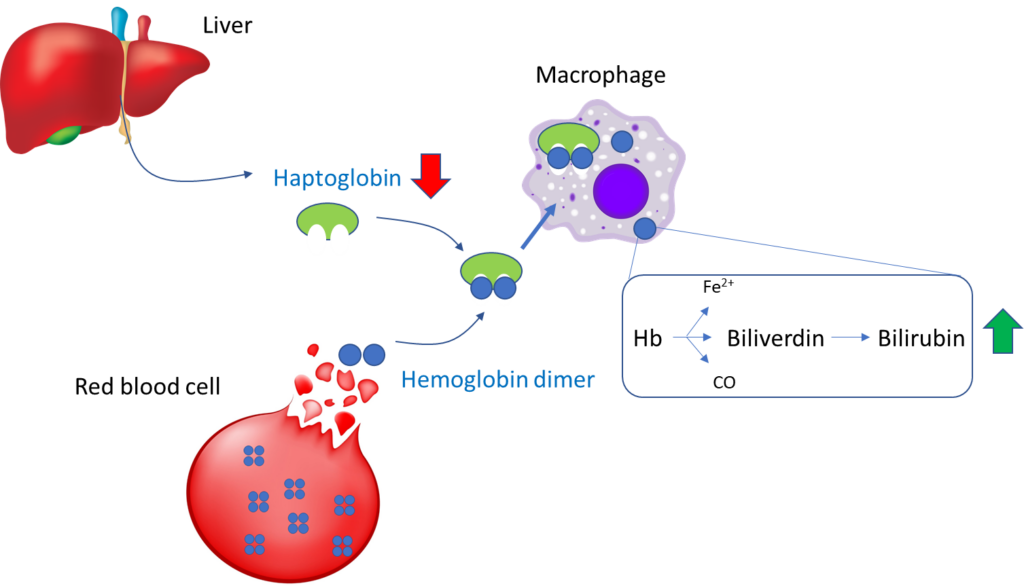
To reiterate, the following are considered serum markers of hemolysis:
- Serum LDH
- Serum bilirubin
- Serum AST
- Serum free Hb
- Serum haptoglobin
In fact, one study showed that mature erythrocytes contain 2,289 distinct gene products! Learn more here.

You will probably agree that of the three parameters (red cell count, hemoglobin and hematocrit), the red cell count is the least useful measure of anemia. One of the reasons for this is that you could have all the red cells in the world, but if they are deficient in hemoglobin and/or smaller than normal in size, the oxygen carrying capacity of blood (which is a function of the hemoglobin concentration) may be impaired.
The same is true when thinking about hemoglobin vs. hematocrit (Hct). Hemoglobin carries oxygen. Hct is simply the fractional volume of blood that is comprised of red cells. It doesn’t tell us anything about hemoglobin levels and oxygen carrying capacity of the blood. In theory, red cells could be completely devoid of hemoglobin, yet maintain a normal Hct.
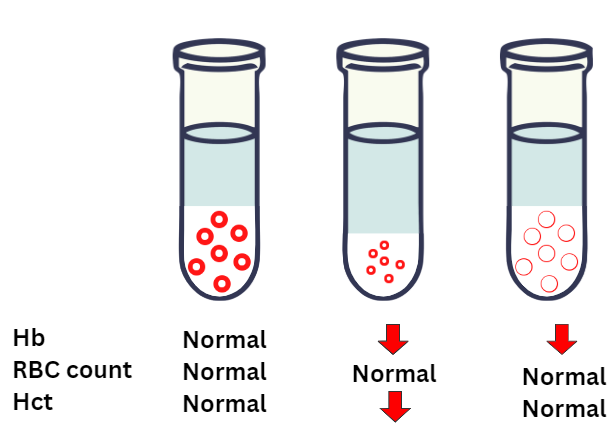
All of this is to say that we want to judge the severity of the patient’s anemia by examining her hemoglobin!
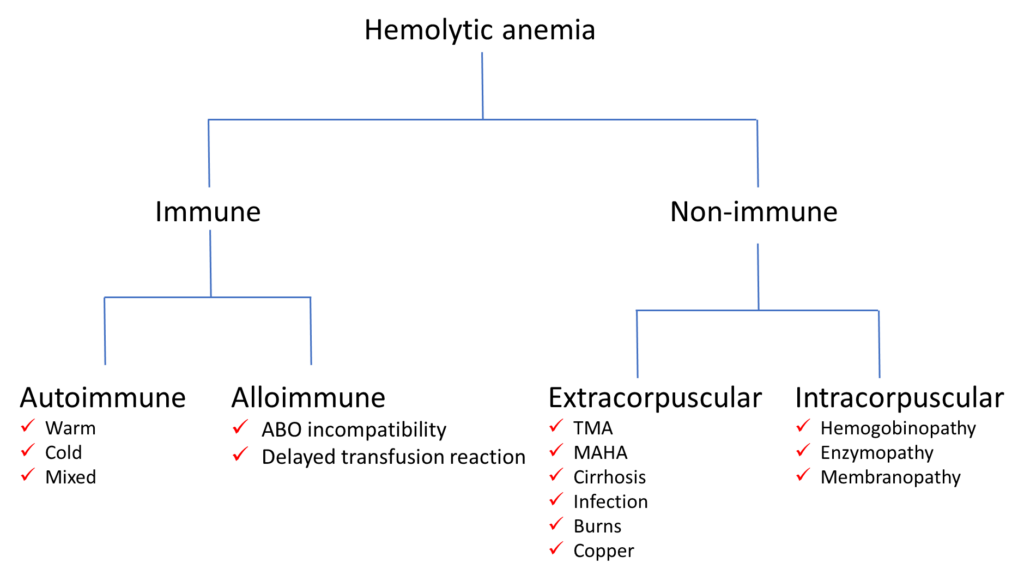
Differential diagnosis of hemolytic anemia
As you prepare to see the patient, it pays to have a broad outline of the differential diagnosis. Nothing too fancy or detailed. Something along the lines of the scheme shown below. We will discuss some of these conditions in a subsequent section when we consider the patient’s lab results.