About the Condition
Description/definition:
Hereditary hemochromatosis (HH) is defined as an inherited iron overload disorder characterized by excessive absorption of iron, due to deficiency of hepcidin.
There are multiple forms of hereditary hemochromatosis, but the most common type is due to a C282Y mutation on the HFE (“high Fe”) hemochromatosis gene. Other gene mutations include mutations in the HJV gene, HAMP gene, TFR2 gene, and SLC40A1 gene.
Classification of hereditary hemochromatosis
- Mutations of HFE gene alleles (classic/type 1 hemochromatosis):
- Autosomal recessive condition.
- Caused by:
- Homozygous C282Y mutation of HFE gene (type 1A):
- G to A transition at nucleotide 845 of the HFE gene.
- Results in a cysteine to tyrosine substitution at amino acid 282 (C282Y).
- Compound heterozygous H63D and C282Y mutation (type 1B):
- May have increased iron indices including transferrin iron saturation and serum ferritin level.
- Penetrance for developing clinically significant iron overload is rare among patients with this genotype (0.5%–2%).
- A third HFE genotype, known as type 1C, is related to the mutation S65C:
- May lead to increased serum iron and ferritin levels.
- Not associated with excess tissue iron stores.
- Can be considered a polymorphism without clinical significance.
- Homozygous C282Y mutation of HFE gene (type 1A):
- Non-HFE-associated hemochromatosis (nonclassical hemochromatosis):
- Account for < 5 % of cases of hereditary hemochromatosis.
- Types 2-4
- Include mutations in:
- Hemojuvelin (HJV gene)
- Hepcidin (HAMP gene)
- Transferrin receptor-2 (TFR2 gene)
- Ferroportin (SLC40A1 gene)
Prevalence in United States, Europe, and Australia, of approximately 1 case in 200–400 persons:
- Highest prevalence exists in people of Irish and Scandinavian origin.
- Lowest prevalence is among those of African descent and Asians.
Over time, iron deposition can lead to dysfunction and failure in multiple organs including the liver, pancreas, heart, joints, and pituitary gland.
The primary goal in the management of HH is to identify patients before end-organ injury and initiate treatment via iron depletion before irreversible end-organ damage develops.

Pathophysiology:
Iron homeostasis
Body iron stores are regulated at the level of intestinal iron absorption, as there are no physiologic processes for the excretion of excess iron other than blood loss via menses or sloughing of senescent intestinal mucosal or epidermal cells.
Hepcidin:
- Considered the key regulator of iron stores by inhibiting of iron absorption.
- A 25-amino acid peptide produced mainly in the liver.
- Produced in response to circulating iron levels.
- Binds to ferroportin (FPN1 ) on macrophages, intestinal absorptive cells, and other tissue cells, after which FPN1 is internalized and degraded; this results in:
- Reduced iron release from the cells.
- Diminished transfer of iron across the enterocyte.
- Reduced iron mobilization from the macrophage.
HFE gene-associated hemochromatosis:
- Caused by dysfunction of HFE gene protein.
- Leads to hepcidin deficiency (relative to circulating iron and body iron stores), which results in:
- Decreased degradation of ferroportin.
- Increased ferroportin-mediated intestinal absorption of dietary iron into the circulation.
- Increased ferroportin-mediated export of iron via ferroportin from liver and splenic macrophages and hepatocytes into the circulation.
- Increased serum iron leads to increased transferrin saturation and increased uptake and storage of non-transferrin-bound iron by parenchymal cells of the liver, heart and pancreas may lead to oxidative damage to tissues, resulting in fibrosis and cirrhosis, and other end organ damage.
| Hemochromatosis type | Frequency | Gene mutated |
|---|---|---|
| Type 1 | > 95% | HFE; homozygous at position 282 (C282Y) (type 1A), compound heterozygous at positions H63D and C282Y (type 1B) |
| Type 2 | < 5% | Hemojuvulin (HJV gene, type 2a) or hepcidin (HAMP gene, type 2b) |
| Type 3 | < 5% | Transferrin receptor-2 (TFR2 gene) |
| Type 4 | < 5% | Ferroportin (SLC40A1 gene) |
Clinical presentation:
Patients may present:
- Incidentally with abnormal liver function in routine screening.
- With positive family history, prompting screening.
- With symptoms of target organ damage.
Symptoms and signs, if present, may include those related to:
- Arthropathy:
- Typically involving the second and third metacarpophalangeal joints.
- The presentation of HH-associated arthritis is similar to that of osteoarthritis and calcium pyrophosphate deposition disease (CPPD).
- Skin pigmentation:
- Hyperpigmentation may be one of the earlier signs of HH.
- Iron deposits in the skin lead to increased melanin production and deposition, giving rise to the characteristic metallic or slate gray hue commonly referred to as bronzing.
- Usually generalized but frequently is deeper on the face, neck, extensor aspects of the lower forearms, dorsa of the hands, lower legs, and genital region.
- Liver damage:
- The most commonly affected organ in type 1 HH.
- May present with:
- Asymptomatic elevation of serum aminotransferases.
- Nonspecific right upper quadrant pain.
- Complications of end-stage liver disease.
- Lifetime incidence of cirrhosis approaches 10% among untreated men with HH.
- Patients with cirrhosis at risk of developing hepatocellular carcinoma (HCC), which accounts for as much as 45% of deaths in this population.
- Endocrine dysfunction, including:
- Diabetes mellitus:
- Prevalence of diabetes among patients with HH is estimated at approximately 13%–23%.
- Causes include iron-mediated injury of pancreatic beta-islet cells and development of hepatic insulin resistance from associated liver injury.
- Hypogonadism:
- Hypogonadotropic hypogonadism is the most common nondiabetic endocrine disorder in HH.
- Results from iron accumulation in the pituitary gland.
- In men, it presents as impotence, loss of libido, and osteoporosis.
- In women, it causes amenorrhea or, less commonly, premature menopause.
- Hypothyroidism
- Diabetes mellitus:
- Cardiomyopathy:
- Second leading cause of mortality in patients with type 1 HH.
- Accumulation of iron in the heart can result in both restrictive and dilated cardiomyopathy, and arrhythmias including sinus syndrome and atrial fibrillation.
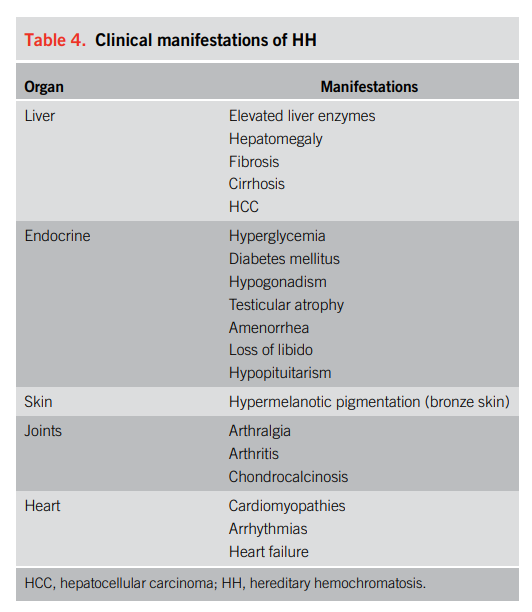
Fatigue and arthralgias are the most common symptoms encountered early in the disease. Up to 18% of men and 5% of women may have hepatic iron overload in the absence of clinical symptoms.
Compared with men C282Y homozygotes, women C282Y homozygotes:
- Present later in life, usually after postmenopause, due to iron loss during menstruation, pregnancy, and lactation offsetting the increased absorption of iron during this time.
- Manifest less commonly with symptoms related to tissue iron deposition; in patients homozygous for C282Y genotype, 25% of men reported to have disease manifestations compared to only 1% of women (thought to be due to protective effects of menstrual blood loss and maternal iron loss during pregnancy).
Diagnosis:
- Consider the diagnosis of hemochromatosis if transferrin saturation (TSAT) > 45% and/or elevated ferritin:
- TSAT is the preferred initial screening test, and fasting is not required to accurately determine TS.
- A TSAT of greater than 45% identifies 97.9%–100% of C282Y homozygotes.
- Serum ferritin is an excellent predictor of advanced fibrosis but lacks specificity as a screening test because hyperferritinemia can be present in other conditions including:
- Alcoholic liver disease
- HCV
- Nonalcoholic fatty liver disease
- Neoplastic disease
- A normal serum ferritin, defined as less than 200 ng/mL in premenopausal women or 300 ng/mL in men and postmenopausal women, in combination with a TSAT of < 45%, has a negative predictive value of 97% for excluding iron overload.
- Confirm the diagnosis of hereditary hemochromatosis with genotype analysis showing C282Y homozygosity or C282Y/H63D heterozygosity.
Guideline recommendations:
Initial testing to identify iron overload:
- Evaluate all patients with evidence of liver disease for hemochromatosis.
- Obtain combination of liver chemistries (LFTs), complete blood count and iron indices (TSAT and ferritin) in patients with suggestive:
- History
- Physical findings
- Family history
Perform HFE mutation analysis if:
- Transferrin saturation ≥ 45% or serum ferritin above upper limit of normal (AASLD).
- Unexplained transferrin saturation > 50% and serum ferritin > 300 mcg/L (for males) or TSAT > 40% and SF > 200 mcg/L (for females) (BSH).
- Individuals have family members (especially first degree relatives) with HFE-related hereditary hemochromatosis.
Studies to determine end organ damage:
- For patients with confirmed HFE C282Y homozygous alleles:
- Investigate with liver function tests (LFTs).
- If normal LFTs, serum ferritin < 1,000 mcg/L and normal clinical exam, monitor serum ferritin and TSAT annually.
- If abnormal LFTs or serum ferritin > 1,000 micrograms/L refer for fibrosis assessment to hepatologist to exclude cirrhosis. Consider elastography a minimal approach. If cirrhosis identified monitor every 6 months with ultrasound and alpha-fetoprotein testing.
- Magnetic resonance imaging can assess liver iron content noninvasively.
- Liver biopsy no longer routinely needed to confirm hereditary hemochromatosis.
- Monitor patients with cirrhosis and genetic hemochromatosis every 6 months with ultrasound and alpha-fetoprotein testing to screen for hepatocellular carcinoma.
- Refer any patients with genetic hemochromatosis and either elevated serum transaminases or serum ferritin > 1000 microg/L to hepatologist for assessment of fibrosis and possible cirrhosis.
Treatment:
Phlebotomy:
- Initial:
- Treatment should be initiated in C282Y homozygotes with an elevated SF, defined as >300 ng/mL in men and >200 ng/mL in women, along with a TS of > 45%.
- All patients with hereditary hemochromatosis and evidence of iron overload with or without evidence of end-organ damage should have regular phlebotomies (weekly if tolerated).


- Maintenance:
- AASLD goal is to maintain serum ferritin (SF) 50-100 mcg/L.
- EASL guidelines report the advocated standard practice is maintenance of SF between 50-100 mcg/L.
- BSH goal is to maintain normal FBC, TSAT index of < 50%, and SF of < 50 mcg/L.
Diet:
- Avoid iron and vitamin C supplements – minimize alcohol intake – increased alcohol consumption of more than 60 g/d can increase the risk of developing cirrhosis in HH by 9-fold, and excess of 80 g of daily alcohol consumption can significantly reduce survival.
- Avoid raw shellfish which may carry risk of Vibrio vulnificus.
Prognosis:
Normal life span if treated before the onset of cirrhosis or diabetes.
Complications of cirrhosis in 20%.
Phlebotomy may lead to improvement in:
- Heart failure
- Fatigue
- Precirrhotic liver disease
- Diabetes
- Abdominal pain
- Skin pigmentation
- Portal hypertension
Treatment may not reverse:
- Cirrhosis
- Arthropathy
- Gonadal failure
- Risk of hepatocellular carcinoma if cirrhosis already present