About the Condition
Description/definition:
Red cell pyruvate kinase deficiency (PKD) is a rare congenital, non-spherocytic hemolytic anemia transmitted as an autosomal recessive trait and caused by a glycolytic defect that is due to compound heterozygous or homozygous mutations in the PKLR gene.
PKD is the most frequent enzyme abnormality of the glycolytic pathway causing hereditary non-spherocytic hemolytic anemia.
Prevalence 1/20,000 to 1/100,000; high frequency of PKD among the Pennsylvania Amish community as a result of a founder effect.

Pathophysiology:
There are 2 PK genes that encode 4 PK isoenzymes. PKD is caused by homozygous or compound heterozygous mutations in the PKLR gene. Over 200 mutations have been described.
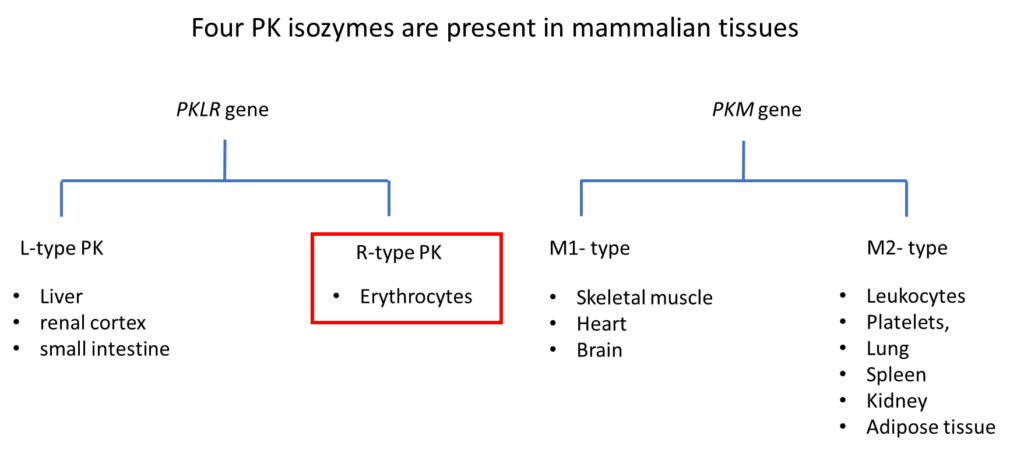
PK catalyzes the conversion of phosphoenolpyruvate to pyruvate in the Emden-Meyerhof (glycolytic) pathway, generating red cell adenosine triphosphate (ATP).
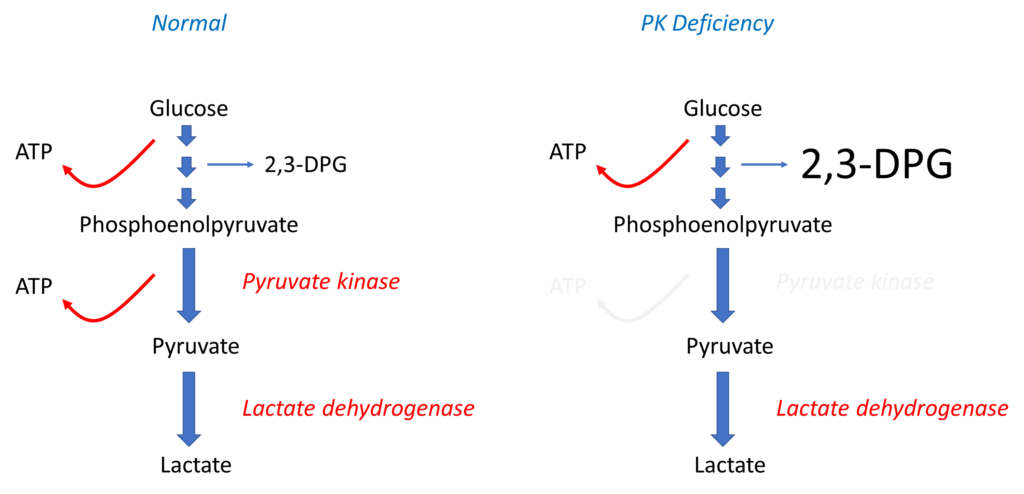
Since mature red cells do not contain mitochondria:
- They rely on glycolysis to generate ATP.
- PK is responsible for generating 50% of the red cell total ATP.
ATP is required for maintenance of red cell functions. In PKD, ATP-depleted red cells:
- Lose large amounts of potassium and water.
- Become dehydrated, rigid and poorly deformable.
- Become entrapped and prematurely destroyed in the the spleen.
Young PK-deficient erythrocytes are most severely affected because of increased ATP requirement of younger cells. While reticulocytes may generate ATP through oxidative phosphorylation not available to mature red cells (because they have mitochondria), oxidative phosphorylation is inhibited by the acidotic, hypoxic and low-glucose environment of the spleen. This may explain the paradoxical finding of increased reticulocyte counts following splenectomy on patients with PKD.
PK deficiency also results in the accumulation of glycolytic intermediates proximal to the metabolic block, including 2, 3-diphosphoglycerate (2, 3-DPG), which causes rightward shift of the oxygen dissociation curve by decreasing Hb affinity to oxygen, thus increasing oxygen delivery to the tissues. This may explain the relatively well-tolerated anemia in some patients with PKD.

From an evolutionary standpoint, PK deficiency has been shown to have a protective effect against replication of the malaria parasite in mouse and human red cells.
Clinical presentation:
The clinical presentation of PKD is highly variable and ranges from intrauterine demise to very mild anemia or fully compensated hemolysis; thus, patients may present any time from very early in life to in young adulthood.
Most patients receive regular transfusions in early childhood, and many patients have a splenectomy at some point in the course of their disease. They may present with complications of ineffective erythropoiesis, hemolysis, splenectomy or transfusion.
Patients may present with complications of
- Ineffective erythropoiesis:
- Frontal bossing
- Spinal cord compression by extramedullary hematopoietic tissue
- Hemolysis:
- Severe anemia
- Gallstones
- Gallstones are detected with increased frequency after the first decade of life and may occur even after splenectomy.
- Aplastic crisis following parvovirus infection
- Chronic leg ulcers
- Splenectomy:
- Overwhelming sepsis
- Thromboembolic events
- Iron overload:
- Heart failure
- Conduction disturbances
- Hypogonadism
- Hypothyroidism
- Hyperpigmentation
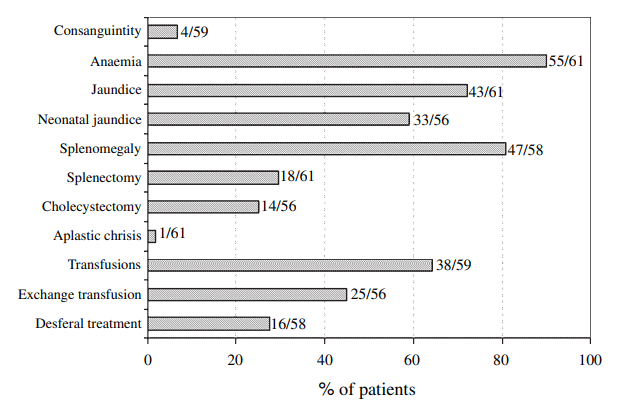
Diagnosis:
Suspect diagnosis of PK deficiency in:
- Patients with evidence of chronic hemolysis in which an immune-mediated hemolytic process, red cell membrane defect, unstable hemoglobin, or paroxysmal nocturnal hemoglobinuria have been excluded.
- Patients who are transfusion-dependent without obvious etiology.
- Neonates with unexplained severe hyperbilirubinemia.
- Patients with undiagnosed hemolytic process in whom reticulocytosis increases after splenectomy, even as the anemia improves.
- Patients with undiagnosed hemolysis and a family history of PKD.
Features of chronic hemolysis include:
- Increased reticulocyte count
- Increased lactate dehydrogenase (LDH)
- Reduced haptoglobin
- Elevated bilirubin
Confirm diagnosis with:
- Direct quantitative enzyme activity (gold standard for the initial diagnosis)
- Molecular testing using next generation sequencing (NGS) strategies
Treatment:
Treatment is mostly supportive.
Transfusions:
- The need for transfusions should be based on comprehensive clinical judgment regarding quality of life, growth, and symptoms and not on hemoglobin level alone.
- Older patients have a lower frequency of transfusions likely linked to the relationship between age and splenectomy and the effect of splenectomy on decreasing transfusion requirement.
Folic acid supplementation:
- Especially important during hemolytic crises, pregnancy, and in childhood during growth and development.
Splenectomy:
- Indications include:
- Patients with PKD who receive regular transfusions or are severely anemic.
- Patients with massive splenomegaly at risk of spleen rupture due to lifestyle choices.
- Typically deferred until patients are > 5 years of age; 5-18 years: best timing.
- Results in a hemoglobin increase of 1–3 g/dL.
- May reduce or even eliminate transfusion.
- Does not arrest hemolysis completely.
- Associated with higher risk of bacterial infections and venous thrombosis
- Immunizations before and after splenectomy
- Prophylactic anticoagulation can be considered, once safe from a bleeding perspective, immediately post-splenectomy.
- Aspirin for thrombocytosis
- Thromboprophylaxis in adults 60 years of age or younger with additional thrombotic risk factors
- Does not decrease the risk of gallstones in patients with PK deficiency
Iron chelation:
- Ideally use to prevent iron accumulation.
- Initiate DFO if ≥ 1 of:
- ≥ 10-20 transfusions
- serum ferritin > 1,000 ng/mL
PK activators:
- Mitapivat, AG-348
- Allosteric small molecule PK activator.
- Shown to activate wild-type and a wide spectrum of mutated PK enzymes in vitro.
- FDA approved for the treatment of hemolytic anemia in adults with pyruvate kinase (PK) deficiency.
Hematopoietic stem cell transplant or gene therapy:
Indications include:
- Failure of PK activator.
- Failure of splenectomy (if patient has two drastic PKLR variants, SCT and gene therapy could be considered prior to splenectomy).
Monitoring:
| Study | Adults (18 years and over) |
|---|---|
| Complete blood counts, reticulocyte count, and bilirubin | At least annually, more often depending on hemolytic episodes and transfusion needs |
| Serum ferritin and TSAT | Every 3-6 months in transfusion dependent; annually in non-transfusion dependent; every 1-3 months while on chelation |
| Liver iron concentration | Annually in transfusion dependent In non-transfusion dependent, MRI frequency, if available, based on the following: annually if > 5 mg/g, every 5 y if < 5 mg/g. |
| Abdominal US | Right upper quadrant US every 2-3 y or until cholecystectomy. After cholecystectomy, every 2-3 y if evidence of intrahepatic cholestasis |
| Viral hepatitis serology | Annually in transfusion dependent |
| Echocardiogram | Consider if age>30 y, prior to pregnancy, and at any age if concern for cardiac dysfunction and/or pulmonary hypertension |