About the condition

Description/definition:
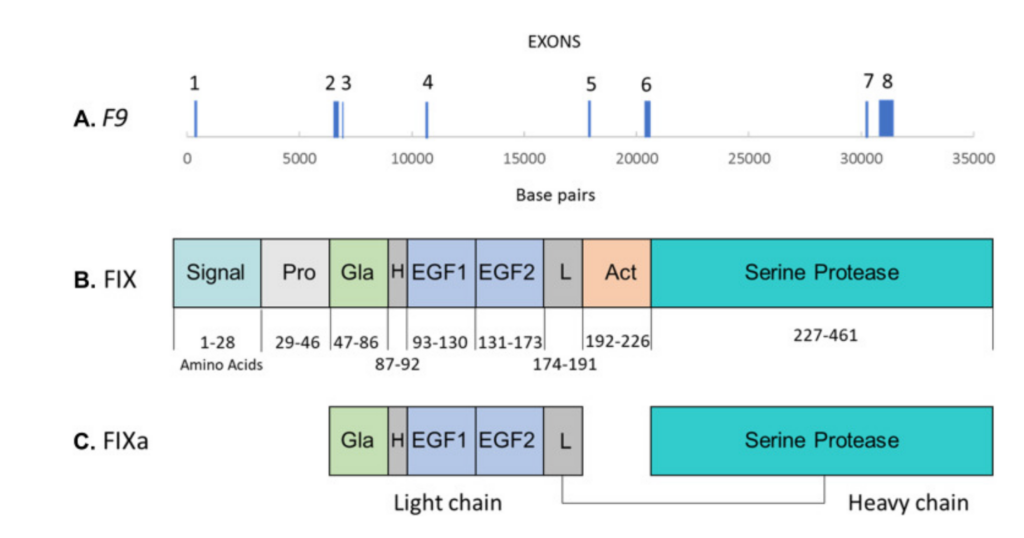
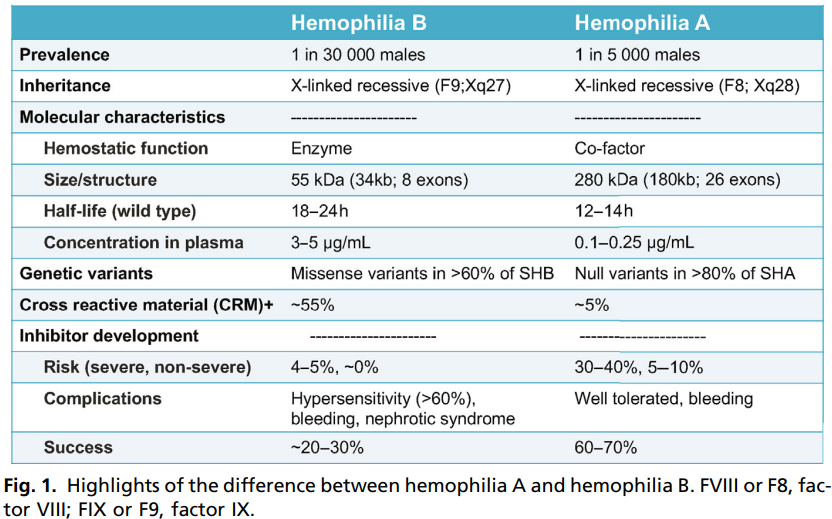
Hemophilia B is a hereditary X-linked chromosomal disorder of varying severity caused by the deficiency or absence of coagulation factor IX (FIX), characterized by spontaneous, postoperative, or posttraumatic bleeding. Reported incidence of hemophilia B is 1 in 20,000 to 1 in 30,000 live male births (accounts for 18% of people with hemophilia)
Classified as:
Mild
- FIX activity > 5%-40%
- Usually diagnosed later in life
- Characterized by prolonged bleeding following major trauma or surgery
Moderate
- FIX activity 1%-5% (0.01-0.05 units/mL)
- Usually diagnosed between age 5 and 6 years
- Characterized by bleeding following minor trauma but may present with spontaneous bleeding
Severe
- FIX activity < 1% (< 0.01 units/mL)
- Usually diagnosed in the first 2 years of life
- May present with spontaneous mild or life-threatening bleeding
A hallmark of hemophilia is bleeding within joints and muscles that in the past often resulted in disability (permanent joint damage)
Pathophysiology:
- FIX is a vitamin K–dependent enzyme that is essential for normal thrombin generation. It is synthesized in the liver.
- Mutations in the F9 gene on the X chromosome, resulting in deficiency or absence of coagulation factor IX (FIX). Over 1000 known pathogenic variants in the FIX gene reported, especially missense and frameshift changes.
- Inherited recessive mutations in about 70% of cases, spontaneous mutations in about 30% of cases (called sporadic cases).
- About 10% of female carriers with 1 F9 disease-causing mutation and 1 normal allele have about 30% factor IX activity and mild bleeding disorder.
- Severity of disease and bleeding complications inversely proportional to factor IX activity
Diagnosis:
Suspect diagnosis of hemophilia A in a patient with:
- Spontaneous bleeding usually into joints, muscles, and soft tissue
- Excessive, prolonged, or intermittent bleeding following trauma or surgery, including heel stick, intramuscular injection, venipuncture
- Family history of bleeding
- Intracranial bleeding in absence of major trauma
- Infant with intracranial hemorrhage or bleeding post circumcision
- Prolonged or renewed bleeding after tooth extraction, circumcision, or mouth injury
- Unexplained gastrointestinal bleeding or hematuria
- Prolonged nosebleeds (particularly recurrent and bilateral)
Confirm diagnosis:
- Factor IX activity level in plasma using a one-stage clot-based or chromogenic substrate assay
- genetic testing identifies disease-causing mutations in 99% of individuals with hemophilia B
Coagulation profile testing may include:
- Elevated aPTT – usually prolonged but may be normal in mild hemophilia B
- Reduced FIX activity level – used for definitive diagnosis, classification of disease severity
- FIX inhibitor testing – if aPTT prolonged
- Alloimmune inhibitors may be present in patients who have been previously exposed to FIX concentrates
- Elevated aPTT will not correct if inhibitor against FIX present
- in contrast to factor VIII inhibitors, factor IX inhibitors are not time-dependent
Differential diagnosis
- Other causes of low FIX activity:
- Normal neonates and premature infants
- Vitamin K deficiency at any life stage
- Inherited deficiency of vitamin K-dependent factors (an autosomal disorder)anticoagulant therapy with vitamin K antagonists
- Liver disease
- Compared with hemophilia A
Treatment:
General principles of management:
- The overall goal of hemophilia B treatment involves utilization of exogenous clotting factor concentrates, or other novel therapies (not currently approved), to achieve hemostasis
- The mainstay of therapy for hemophilia B is to replace the deficient clotting factor to:
- Prevent bleeding (called prophylactic treatment)
- Control bleeding (called on-demand or episodic treatment)
- FIX concentrates include
- Plasma-derived
- Recombinant
Treatment of active bleeding:
- Patients with hemophilia should receive immediate care for potential bleeding events with strong consideration of treatment with clotting factor concentrates before any imaging
- In patients without inhibitors
- In patients with major bleeding, the goal is to replace factor IX activity to ≥ 100%
- Factor IX concentrate (plasma derived) 1 unit/kg increases factor IX activity by 1%, but dosing of factor IX varies by type of product due to different recovery rates (the amount of factor IX concentrate that is required to achieve a similar plasma level is approximately twice that of an FVIII product)
- Recombinant human factor VIIa may be given if patient has history of previous allergic reaction to factor IX
- Antifibrinolytic monotherapy or adjuvant therapy dosing for mild mucosal or skin bleeding (for example, during dental surgery)
- In patients with inhibitors
- For low-titer inhibitor patients (< 5 Bethesda units/mL)
- Factor IX concentrate – high-dose or high-frequency with the goal in patients with major bleeding to replace factor IX activity to ≥ 100%
- For high-titer inhibitor patients (> 5 Bethesda units/mL)
- Recombinant hFVIIa or APCC (FEIBA)
- Antifibrinolytic monotherapy (tranexamic acid or aminocaproic acid) for mucosal or skin bleeding
- For low-titer inhibitor patients (< 5 Bethesda units/mL)
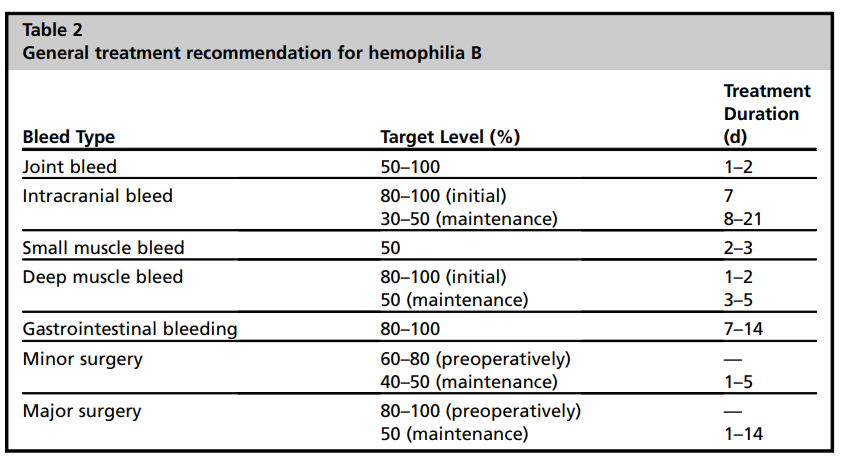
Prophylactic treatment
- Considered standard of care in all patients with severe hemophilia B
- Historically involves the routine administration of factor IX concentrate to maintain at least a 1% trough FIX activity in between dosing
- For patients without inhibitors, consider factor IX – typical initial dosing 15-40 units/kg IV twice weekly; regimens are often individually tailored
- For patients with inhibitors (> 5 Bethesda units/mL) consider either:
- factor VIIa (recombinant) – typical dosing 90 mcg/kg IV given once daily
- aPCC – typical dosing 50-85 units/kg IV every other day or 3 times weekly
- if treatment failure occurs, consider giving the alternate agent