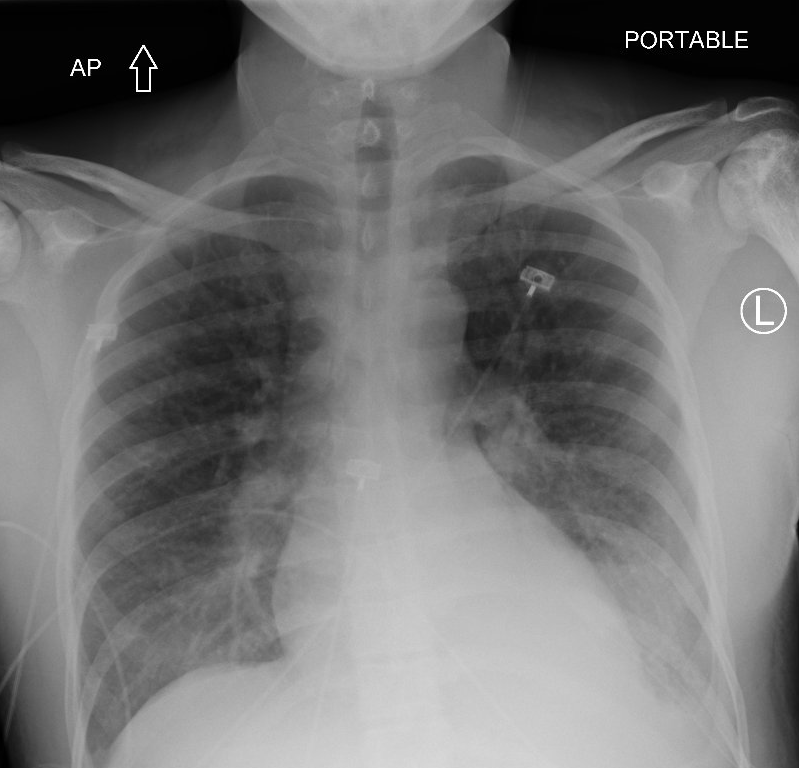
Imaging and Labs

The following is a partial complete blood count (CBC) on the day of admission and her baseline CBC for comparison:
| Day | WBC (109/L) | Hb (g/dL) | MCV (fL) | PLT (109/L) |
|---|---|---|---|---|
| Day of transfer | 15.4 | 6.9 | 75 | 92 |
| 6 months prior | 8.1 | 12.3 | 73 | 155 |
What’s what: WBC, white blood cell count; Hb, hemoglobin; MCV, mean cell volume; MCHC, mean cellular hemoglobin concentration; RDW-SD, red cell distribution width-standard deviation; platelets, PLT; Normal values: WBC 5-10 x 109/L, RBC 4-6 x 1012/L, Hb 12-16 g/dL, Hct 35-47%, MCV 80-100 fL, MCHC 32-36 g/dL, RDW-SD < 45%, platelets (PLT) 150-450 x 109/L
The white cell differential:
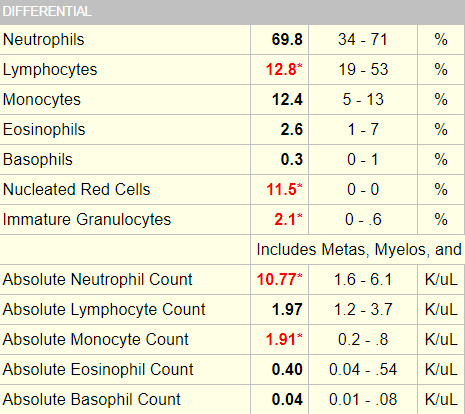

Also note the nucleated red cells, which are characteristic of patients with sickle cell disease.
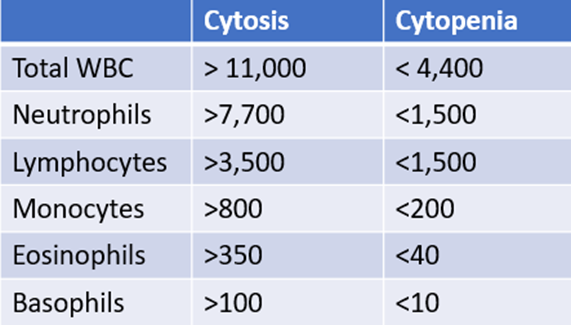
How do you know which white cell values are high or low? Because you carry around the following cheat sheet on your iPhone!
Reticulocyte count on day of admission:
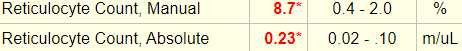
Ignore the percentage! Focus on the absolute reticulocyte count. Absolute reticulocyte count = 0.23 x 1012/L = 230 x 109/L
So, the patient’s reticulocyte count is appropriate. What diagnosis would you entertain if the absolute reticulocyte count was 0.01 x 1012/L?
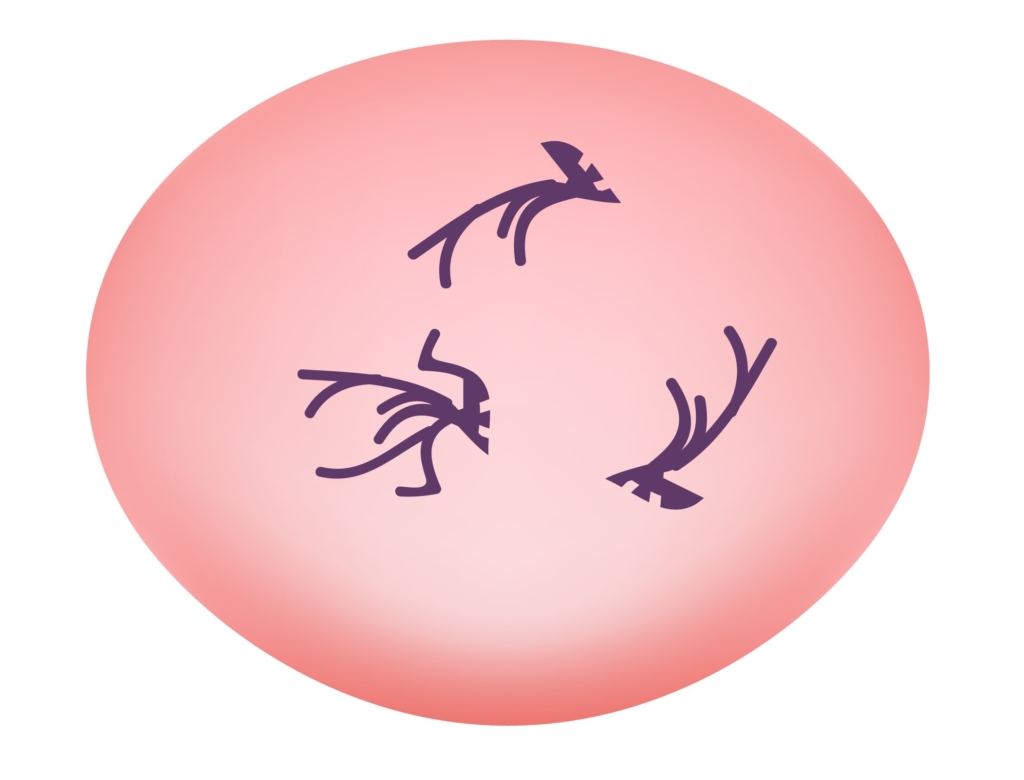
Which peripheral smear is most likely to resemble the patient’s?
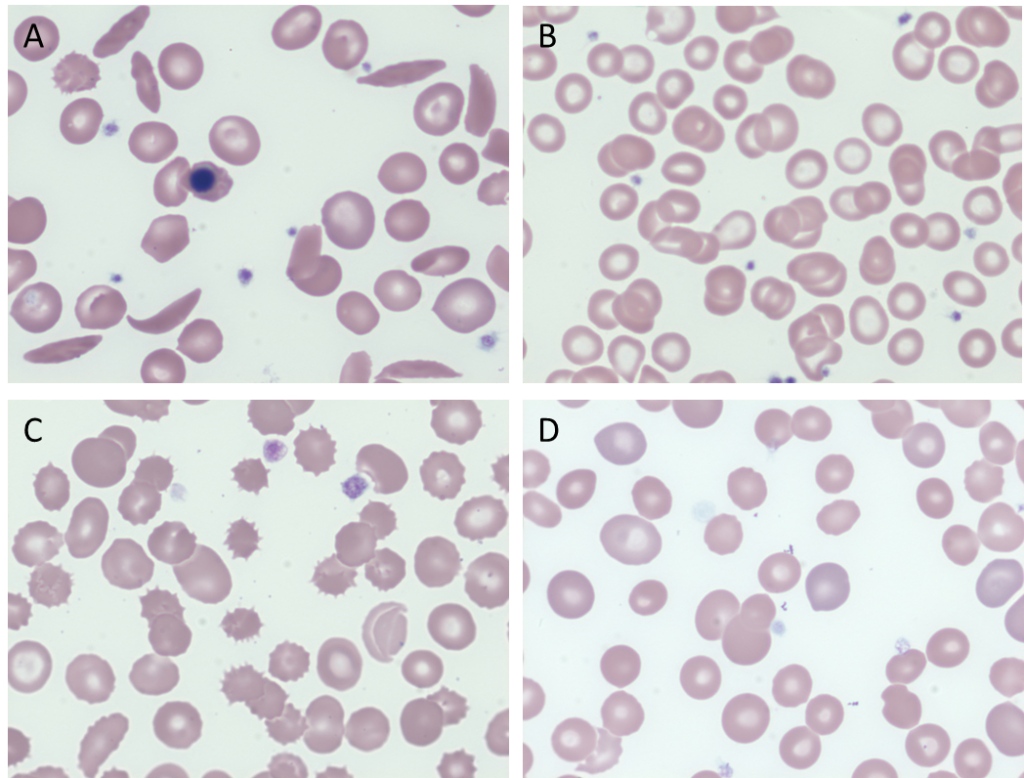
Which blood smear schematic is most consistent with the patient’s?
Let’s pause for a moment and consider genotype-phenotype correlations in sickle cell disease as they pertain to HbSS and HBSC.
Definitions of sickle cell disease and sickle cell anemia
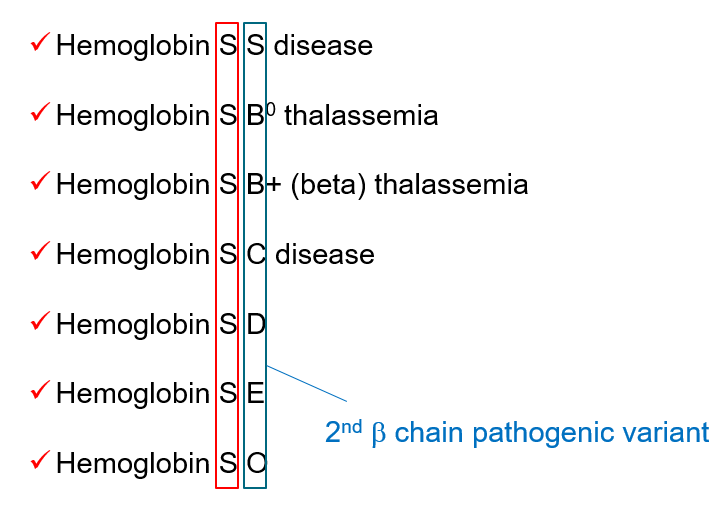
- Sickle cell disease (SCD) refers to a group of disorders characterized by the presence of at least one hemoglobin S allele (HbS; p.Glu6Val in HBB) and a second HBB pathogenic variant resulting in abnormal hemoglobin polymerization. SCD includes:
- Homozygous for HbS (HbSS)
- Sickle beta0 thalassemia (compound heterozygote with one sickle gene and one beta0 thalassemia gene)
- Hemoglobin SC disease (HbSC) (compound heterozygous)
- Sickle beta+ thalassemia (compound heterozygous)
- Rarer forms, including:
- SD-Punjab (also known as HbSD)
- SO-Arab
- SC-Harlem
- S-delta-beta-thalassemia
- SE
- S-Lepore
- Sickle cell anemia is a subset of sickle cell disease that refers to individuals who are either of the following:
- Homozygous for HbS (HbSS)
- Sickle beta0 thalassemia (compound heterozygote with one sickle gene and one beta0 thalassemia gene)


Hemoglobin SC
- A genotype of sickle cell disease (SCD).
- Caused by co-inheritance of the hemoglobin S (HbS) and hemoglobin C (HbC) beta globin gene (HBB) mutations.
- Accounts for 30% of sickle cell disease (SCD) in the United States.
- Survival of patients with HbSC is superior compared to SCA.
- Pathophysiological processes include:
- Altered K–Cl cotransporter contributing to RBC dehydration, which:
- Increases the intracellular hemoglobin concentration
- Makes the cell more dense than HbAA-containing red cells
- Enhances the pathogenic properties of HbS
- Polymerization of HbS subunits
- Crystallization of HbC subunits
- Altered K–Cl cotransporter contributing to RBC dehydration, which:
- Improved outcome with increased levels of HbF or co-inheritance of alpha thalassemia gene deletions.
- Compared with patients with HbSS, those with HbSC have:
- Higher mean Hb
- >70% of people with HbSC have anemia
- Only 10% have Hb lower than 10 g/dL
- Lower mean corpuscular volume (MCV) (inheritance of alpha thalassemia trait further reduces MCV)
- Increased MCHC
- In some but not all patients
- Average MCHC in HbC disease, HbSC disease, HbAC trait, and HbA cells 38, 37, 34, and 33 g/dl, respectively
- Higher mean Hb
- Lower absolute reticulocyte count
- 2-fold greater red cell life span
- Increased blood viscosity
- Increased incidence of
- Proliferative sickle retinopathy
- Acute splenic sequestration crisis (chronic splenomegaly is associated with thrombocytopenia in 35% of children and 50% of adults with HbSC and may cause recurrent abdominal pain)
- Peripheral blood smear may show:
- Target cells
- Irregularly contracted cells
- Boat shaped cells (but few classic sickle cells)
- Microcytic and hyperchromic red cells
- Microspherocytes
- Red cells with crystalline inclusions
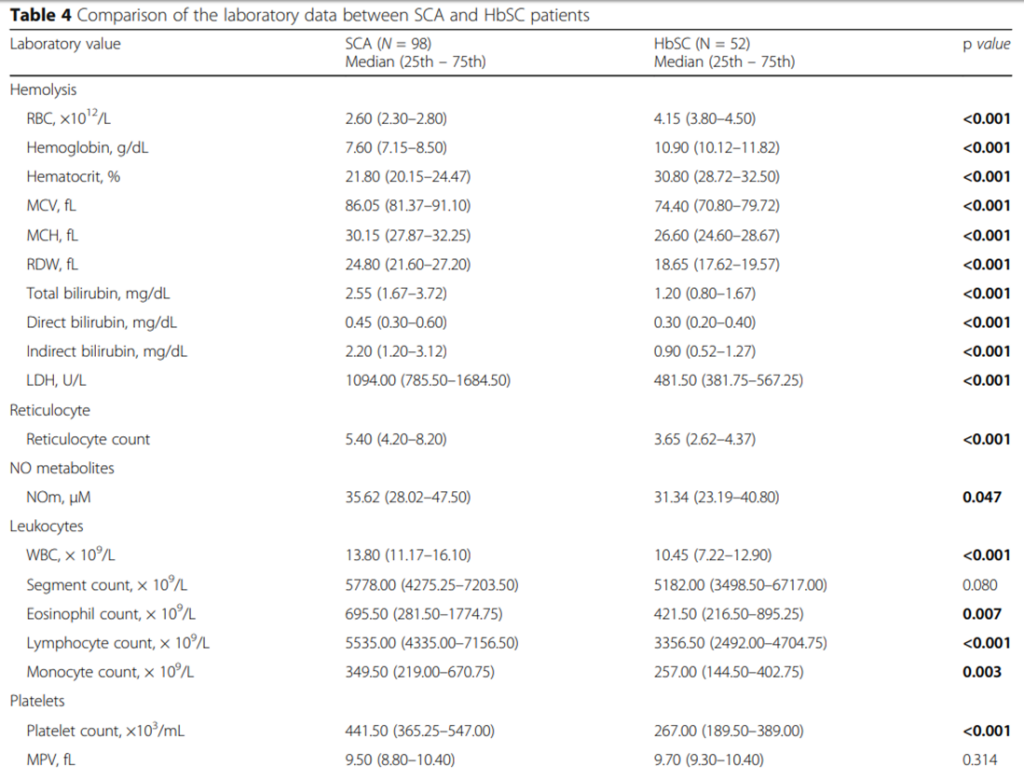
The following are complete blood count data and hemolysis markers comparing HbSC and HbSS:

Liver function tests, urea and creatinine were normal in our patient.
The following are the hemolysis labs:
| Baseline (1 month earlier) | Day of transfer | |
|---|---|---|
| LDH (IU/L) | 203 | 1461 |
| AST (IU/L) | 14 | 73 |
| ALT (IU/L) | 8 | 12 |
| Haptoglobin (mg/dL) | 44 | <10 |
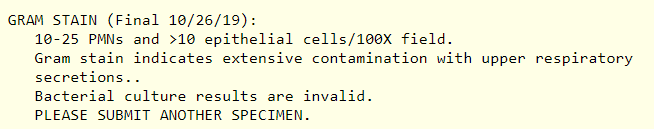
Gram stain sputum:
Arterial blood gas was not performed.