Abstract
Blood is essential for many biological functions in humans, most notably oxygen and nutrient delivery to cells. Millions of red blood cell concentrates are transfused every year to treat anemia and acute blood losses. In addition to their therapeutic uses, blood transfusions are also employed for performance enhancement in professional sports, especially in endurance disciplines, where improved oxygen delivery capacity can provide a distinct competitive advantage. The detection of autologous blood transfusion use remains a considerable analytical challenge for anti-doping laboratories. Blood is not only a potent performance-enhancing agent, but is also an important biological matrix for the detection of numerous drugs in elite athletes. In this narrative review, the significance of blood for sports drug testing will be discussed – both regarding blood doping and as indispensable sample matrix.
Abbreviations
AAF = Adverse analytical finding
AAS = Anabolic androgenic steroid
ABP = Athlete Biological Passport
ABPS = Abnormal Blood Profile Score
ActRIIA/B = Activin receptor type IIA or B
ADAMS = Anti-Doping Administration and Management System
APMU = ABP Management Unit
ATP = Adenosine triphosphate
BCO = Blood collection officer
BHK = Baby hamster kidney
CE = Capillary electrophoresis
CERA = Continuous EPO receptor activator
CHO = Chinese hamster ovary
CLIA = Chemiluminescent immunoassay
CO = Carbon monoxide
DBBF = Dibromosalicyl fumarate
DCO = Doping control officer
del/del = deletion/deletion
DEHP = di-(2-ethylhexyl)phthalate
2,3-DPG = 2,3-diphosphoglycerate
DPS = Dried plasma spots
E = Epitestosterone
EPO = Erythropoietin
ERA = Epo receptor agonist
ESAs = Erythropoiesis-stimulating agents
Fc = Fragment crystallizable
FIS = Fédération Internationale de International Ski
GC = Gas chromatography
HBOCs = Hemoglobin-based oxygen carriers
HCD = Higher energy collision dissociation
Hct = Hematocrit
hGH = Human growth hormone
HIF-1α = Hypoxia-inducible factor 1 α
HRMS = High-resolution mass spectrometry
IAAF = lnternational Amateur Athletic Federation
IEF = Isoelectric focusing
IGF-I = Insulin-like growth-factor I
IgG = Immunoglobulin G
IOC = International Olympic Committee
IRMA = Immunoradiometric assay
IRMS = Isotope ratio mass spectrometry
ISL = International Standard for Laboratories
LC = Liquid chromatography
LLE = Liquid-liquid-extraction
%Macro = Percent macrocytes
MDGC = Multi-dimensional gas chromatography
MS = Mass spectrometry
MS/MS = Tandem mass spectrometry
NESP = Novel erythropoiesis-stimulating protein
N-FID = Nitrogen flame ionization detector
NIR = Near-infrared
NO = Nitric oxide
PEG = Polyethylene glycol
pI = Isoelectric point
P-III-NP = Procollagen type III
PVC = Polyvinyl chloride
RBC = Red blood cell
Ret% = Percent reticulocytes
rEPO = Recombinant EPO
RetHct = Reticulocyte hematocrit
rhEPO = Recombinant human EPO
rhGH = Recombinant hGH
SAR = Sarcosyl
SDS-PAGE = Sodium dodecyl sulfate polyacrylamide gel electrophoresis
SEC = Size-exclusion chromatography
SPE = Solid-phase extraction
sTfr = Soluble transferrin receptor
T = Testosterone
TGF-β = Transforming growth factor beta superfamily
TOF = Time-of-flight
UCI = Union Cycliste Internationale
UHPLC = Ultra-high-performance LC
WADA = World Anti-Doping Agency
WBC = White blood cells
WADC = World Anti-Doping Code
1. Introduction
For almost 50 years, doping controls have been a key element of professional sports to protect values such as honesty, fairness, respect, and health. As shown in Figure 1, doping control samples are collected at the competition or training site of the athlete under constant supervision of a doping control officer (DCO) of the same gender. While the obtained urine is split into A and B specimens and subsequently sealed in the corresponding bottles, two individual A and B blood samples must be collected in the respective blood collection tubes (see also Section 3).
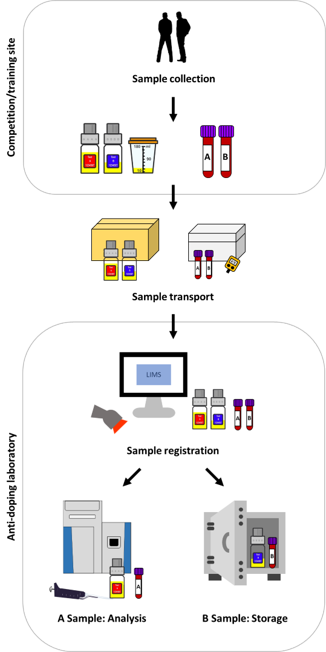
Both urine and blood specimens are labeled with a unique code number and anonymously sent to a laboratory accredited by the World Anti-Doping Agency (WADA). Following arrival, samples are registered, and the A specimen is tested for the presence of prohibited doping agents, their metabolites, or biomarkers indicative for doping, mostly by using mass spectrometric and/or immunological approaches. The analytical results are sent to the responsible anti-doping organization or sports federation, which possesses the information to decode the sample and inform the athlete of the test result. The corresponding B sample is securely stored and only analyzed upon request in case of an adverse analytical finding (AAF). In 2019, a total of 278,047 A samples were analyzed by 31 anti-doping laboratories. 1. 90.9% (= 252,708) were urine specimens while only 9.1% (= 25,339) were composed of blood including whole blood, plasma and serum. In 0.97% of these samples, an AAF was obtained and as in previous years, most of these could be attributed to anabolic androgenic steroids (AAS). Additionally, 36,401 whole blood samples for hematological athlete biological passport (ABP) measurements (see also Section 3.1.1) were collected.
Even though blood is less commonly analyzed than urine by anti-doping laboratories, the use of blood specimens is indispensable because some doping agents are more readily (and in some cases exclusively) detected in this sample matrix. Blood is not only a valuable biological matrix for sports drug testing, but is also a potent doping agent itself. About 5 liters of blood circulate in the body of a human adult and the most important functions of this body fluid are the transport of oxygen, nutrients (glucose, amino acids, and fatty acids), electrolytes, hormones, and metabolic waste products (e.g., carbon dioxide and urea; immunological defense; and coagulation.2 Many of these functions are fulfilled by different blood cells derived from the bone marrow, which constitute approximately 45% of the blood volume and comprise red blood cells (RBCs), several types of white blood cells (WBCs), and platelets. While RBCs (also called erythrocytes, 3.5-5.9 x 1012/L of blood) are responsible for the transport of oxygen to the cells of the body, WBCs (also called leukocytes, 4.5-11.0 x 109/L of blood) including lymphocytes, granulocytes, and monocytes are part of the immune response. Finally, platelets (also called thrombocytes, 150-400 x 109/L of blood) are cellular fragments forming blood clots following activation to prevent loss of blood from the vasculature. Blood cells are suspended in plasma, a liquid composed of water, different proteins (mainly serum albumin, antibodies, and coagulation factors), nutrients, electrolytes, and waste products.
Due to their pivotal role in oxygen transport, an artificially or naturally increased number of circulating RBCs can result in an improved athletic endurance performance.3 However, blood doping still represents an enormous analytical challenge for anti-doping laboratories on the one hand and requires a considerable logistic and practical effort for the cheating athlete (and the medical supporter) on the other hand. In this review, the importance of whole blood, plasma, and serum as biological matrices for the detection of different doping agents is discussed. Moreover, the role of blood as doping agent is comprehensively described.
2. Past
At the beginning of systematic sports drug testing programs during the 1970’s, urine was the preferred biological matrix for the detection of performance-enhancing drugs as it involved a simple, non-invasive sample collection, and the excreted concentrations of the most relevant doping agents were compliant with the sensitivity of early analytical procedures.4 At that time, the list of prohibited substances published by the medical commission of the International Olympic Committee (IOC) included stimulants and narcotics (both added in 1967), and AAS (added in 1976),5 which were detected in urine by using gas chromatography (GC) in combination with a nitrogen-specific flame ionization detector (N-FID) or mass spectrometry (MS).6
However, with the increasing misuse of recombinantly produced peptide hormones such as human growth hormone (hGH) and blood doping, the use of whole blood/serum/plasma for doping control analysis became inevitable.7 Due to the size and charge selectivity of the glomerular filter as well as reabsorption processes in the renal tubules, urinary concentrations of high-molecular-mass compounds are usually very low. Therefore, blood is the preferred biological matrix for the detection of such analytes. Moreover, it represents the only measure to provide evidence for homologous and autologous blood doping by blood group analysis and the evaluation of relevant hematological parameters (see also Section 3). The high resistance of blood as sample matrix towards manipulation by cheating athletes (e.g., by sample substitution or adding proteases) as well as the possibility to assign a sample to an athlete by genetic fingerprinting were considered as further advantages of blood sample analysis.
At the same time, the introduction of blood sampling to sports drug testing was associated with several ethical, legal, and practical considerations 8: First, the sample collection procedure is invasive and needs to be conducted by trained medical personnel to minimize the risk of physical injury and disease transmission.9 Second, compared to urine, blood is not a “waste product”, and the available sample volume is restricted. Third, blood concentrations of “classical” doping agents and their metabolites are significantly lower than in urine, and as the established analytical “gold standard” GC-MS is not applicable to the detection of peptide hormones and blood doping, the development of novel validated direct and/or indirect assays was required. Finally, strategies had to be put in place to ensure uniformly high standards for blood sample collection, transportation, storage, and analysis.
Doping control blood specimens were first collected by the International Skiing Federation (Fédération Internationale de Ski, FIS) at the World Championships in Lahti, Finland in 1989.10 Here, a total of 66 samples were tested for homologous blood doping by blood group analysis.11 Moreover, hemoglobin (Hb), hematocrit (Hct), and erythropoietin (EPO) levels were determined and compared to population mean data. All values were found to be within normal ranges and no blood doping was detected. In the following years, blood sampling was routinely conducted by the FIS at championships and major World Cup events,12 and in 1993/94, also the lnternational Amateur Athletic Federation (IAAF) collected whole blood and serum specimens from 99 athletes at eight World Cup meetings.13 These samples were analyzed for homologous blood doping, and different blood parameters as well as endogenous hormone and binding protein levels were determined to uncover hGH, EPO, and testosterone misuse. All tests returned negative, which may, to some extent, be attributed to an insufficient sensitivity and/or specificity of the employed assays.
In 1993, the IOC’s medical commission made a recommendation to introduce blood tests at the Olympic Games, which were carried out along with urine sampling on FIS athletes during the Olympic Winter Games 1994 in Lillehammer, Norway.14 FIS-approved guidelines for sample collection were employed and samples were tested for homologous blood transfusions only. Shortly thereafter, the meanwhile acquired medical data was employed by the FIS to introduce thresholds of 16.5 g/dl for female and 18.5 g/dl for male skiers to exclude athletes with higher levels from competing.
In 1997, the International Cycling Union (Union Cycliste Internationale, UCI) started a program of unannounced blood tests conducted at the site of competition to protect the athlete’s health and support clean sports: The hematocrit was measured and athletes exceeding thresholds of 50% (men) and 47% (women) were not allowed to participate in competitions for 15 days.15 During the Olympic Games 2000 in Sydney, comprehensive blood sampling was conducted for the first time.16 More than 300 athletes provided paired blood and urine samples,17 which were tested for the misuse of recombinant human EPO (rhEPO) by using direct (urine),18 and indirect (blood) approaches.19 While the five-parameter “ON” model for the identification of current EPO misuse based on the analysis of hematological parameters yielded no positive findings, the three-parameter “OFF” test for recent EPO doping returned suspicious results for seven athletes, which warranted further follow-up such as unannounced out-of-competition testing.20 And with the establishment of the WADA in 1999 and the implementation of the World Anti-Doping Code (WADC) by numerous sports organizations in 2004, the way for internationally harmonized and coordinated anti-doping programs including both urine and blood samples was finally paved.21
3. Present
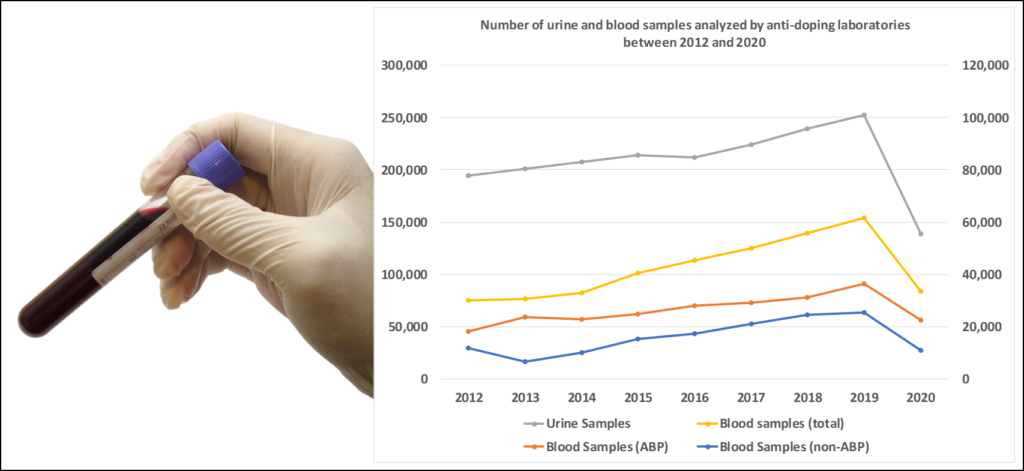
Nowadays, both blood and urine samples are routinely analyzed by WADA-accredited anti-doping laboratories, and as shown in Figure 2, the total number of blood samples reported into the Anti-Doping Administration and Management System (ADAMS) more than doubled from 2012 (30,221) to 2019 (61,740).22 Every year, approximately 60% of the blood specimens constitute whole blood for hematological ABP measurements, and the remaining samples are serum, plasma and whole blood for the detection of peptide hormones, homologous blood transfusions, and selected other doping agents (see below).
According to current WADA guidelines,23 very strict rules apply to the collection, transportation, storage, and analysis of doping control blood samples in order to ensure maximum levels of quality for sample analysis and a high inter-laboratory comparability. To protect the health of both the athletes and sample collection personnel, venipuncture has to be conducted by a qualified blood collection officer (BCO). Here, the non-dominant arm should be used to affect the respective athlete and its performance as little as possible. In case of unexpected problems during venipuncture, a maximum of three attempts is allowed. The required amount and type of blood (whole blood/plasma/serum) as well as the total number of samples depend on the analytical requirements: While hematological measurements and testing for homologous blood transfusions necessitate non-coagulated whole blood, only serum can be used for the determination of hGH isoforms and biomarkers. For the analysis of erythropoiesis-stimulating agents (ESAs), hemoglobin-based oxygen carriers (HBOCs), and steroid esters, both serum and plasma can be employed. Except for ABP samples, two tubes representing A- & B-sample are mandatory. The collected blood specimens are then sealed in a tamper evident kit and transported to the anti-doping laboratory in refrigerated condition to maintain their integrity. Until sample reception, the temperature in the transportation device has to be continuously monitored by a data logger as both warming and freezing have to be avoided. For ABP samples, the maximum allowed period of time for sample arrival at the laboratory depends on the average temperature during transportation: 60 h when kept at 4°C, and 35 h when transported at 15°C.
Following arrival, samples of which the serum/plasma fraction is used for analytical testing should be centrifuged as soon as possible and subsequently stored frozen.24 Aliquots of the A specimen may also be stored refrigerated for a maximum of 24 h. If sample analysis is performed on the cellular fraction, the blood needs to be maintained refrigerated until all measurements are completed. Then, it can be centrifuged to obtain plasma for additional tests, which has to be stored as described above.
3.1 Blood as doping agent
Especially in endurance disciplines, athletic performance can significantly be improved by increasing the body’s RBC mass and thus the oxygen delivery capacity of the blood.25 To achieve blood manipulation, different substances and techniques can be misused, one of them being the transfusion of RBCs obtained from the athlete him-/herself (autologous) or another person with compatible AB0 and Rhesus (D) blood groups (homologous/allogeneic).26
Even though the beneficial effects of blood transfusions have been recognized since at least the early seventies,27 their use in sports was not banned until 1986, when the confessions of several athletes to have supported endurance performance by blood manipulation resulted in the addition of blood doping to the IOC’s list of prohibited substances.28 With the clinical approval of rhEPO in the late 1980s, the use of blood transfusions became temporarily less attractive for cheating athletes as EPO has not only the same physiological effects but is also easier and safer to administer.29 But after the introduction of a direct detection method for EPO doping in 2001,30 the use of blood transfusions for performance enhancement regained the interest of athletes as their misuse was – at that time – more difficult to detect.31
Irrespective of the blood’s origin (autologous/homologous), the administration of RBC concentrates to healthy volunteers can be associated with severe side effects such as phlebitis, bacterial infections due to an inadequate handling or storage of the blood, and hyperviscosity, which possibly leads to venous thromboembolism.32 Moreover, the “logistics” per se can pose a significant health risk regarding the development of thrombosis, as recent reports suggest that some athletes were even used as “autologous blood body packers” to transport blood bags intended for doping to the competition site. 33 Shortly before a long-distance flight, they were transfused with the required amount of blood, which was re-collected following arrival at the destination and stored refrigerated until the competition. Additionally, there are specific health risks associated with the type of blood doping, which will be discussed in the following sections.
3.1.1 Autologous blood doping
During autologous blood doping, athletes use their own blood following ex vivo storage to increase RBC and Hb mass and thus athletic performance.34 To achieve a significant improvement in the maximal aerobic power (VO2Max), a minimum of two blood units ideally stored frozen (see below) has to be transfused.35 From an athlete’s point of view, the biggest advantage of this approach is that its detection by doping control laboratories is very challenging as the composition of the surface antigens between the transfused and circulating RBCs are identical. Therefore, mostly indirect approaches are employed, the most important one being the hematological module of the ABP (see below). Additionally, the risk for adverse events such as hemolytic reactions due to blood group incompatibility and disease transmission is significantly reduced when compared to homologous blood doping.36
On the downside, this doping method requires a high organizational effort as blood withdrawal, storage, and reinfusion have to be synchronized with competitions and training periods.37 For RBCs separated from other blood components and mixed with anticoagulant and additive solutions, the maximum storage period at 4°C (blood bank conditions) is only 6 weeks.38 During this period, the number of functional RBC continuously declines due to normal ageing of the cells as well as the development of storage lesions (see below), resulting in an overall 24-h-post-transfusion survival rate of only 75%.39 At the same time, physical performance is significantly decreased as it usually takes several weeks until Hb levels are restored following blood sampling.40 This intermittent negative effect can be prevented by a preceding use of EPO to stimulate RBC production prior to venesection, which, however, increases the risk of being caught by anti-doping authorities as the misuse of this peptide hormone can be directly detected in blood and urine specimens (see also 3.2.2). Moreover, athletes have to consider that other doping agents such as anabolic steroids used during training before blood withdrawal can be reintroduced into their body together with the stored blood and result in an AAF.41
Alternatively, RBC can also be mixed with glycerol and stored for several years at –80°C or in liquid nitrogen.42 Freezing under such conditions suspends RBC ageing and only 15% of the cells are lost during the entire handling process. Additionally, the longer storage period gives athletes a greater temporal flexibility. However, this technique is very laborious and expensive as the cells have to be carefully thawed and washed several times to remove the glycerol prior to their reinfusion.
The confessions/statements of several athletes caught for doping as well as confiscated diaries/calendars gave detailed insights into autologous blood doping strategies usually employed in sports: In training periods without competitions, injections with rhEPO are used to artificially increase RBC mass. After 2-3 weeks of treatment, one to three units of blood are withdrawn, stored refrigerated/cryopreserved for several weeks/months, and re-infused a few days prior to a competition. Shortly thereafter, two blood bags are withdrawn again and re-infused shortly before the next tournament and so on.43
3.1.1.1 The athlete biological passport (ABP)
As mentioned above, the detection of autologous blood doping represents an enormous challenge for anti-doping laboratories and organizations, and the most promising indirect approach – which can also reveal other blood doping practices – is the hematological module of the ABP, which was proposed as early as 2000 44 and finally installed in 2009.45 The key concept of the ABP is that performance enhancement due to doping is based on physiological changes, which can remain much longer in the body than the drug itself and individually be assessed for each athlete.46 In the hematological module, different variables and multiparametric markers indicative of blood manipulation such as the Hb concentration, reticulocyte percentage (Ret%), and Abnormal Blood Profile Score (ABPS) are monitored for each athlete over time.47 While initial thresholds for the different biomarkers are population-based, they are individually adapted in the course of data acquisition.48 In addition to the hematological values, also heterogeneous information as the athlete’s age and sex as well as confounding factors such as exposure to altitude, are included and evaluated by experts in cases where the ABP software identifies abnormal blood profiles.49 Those can either be used to directly pursue an anti-doping rule violation or identify athletes for targeted ESA or homologous blood transfusion testing.50
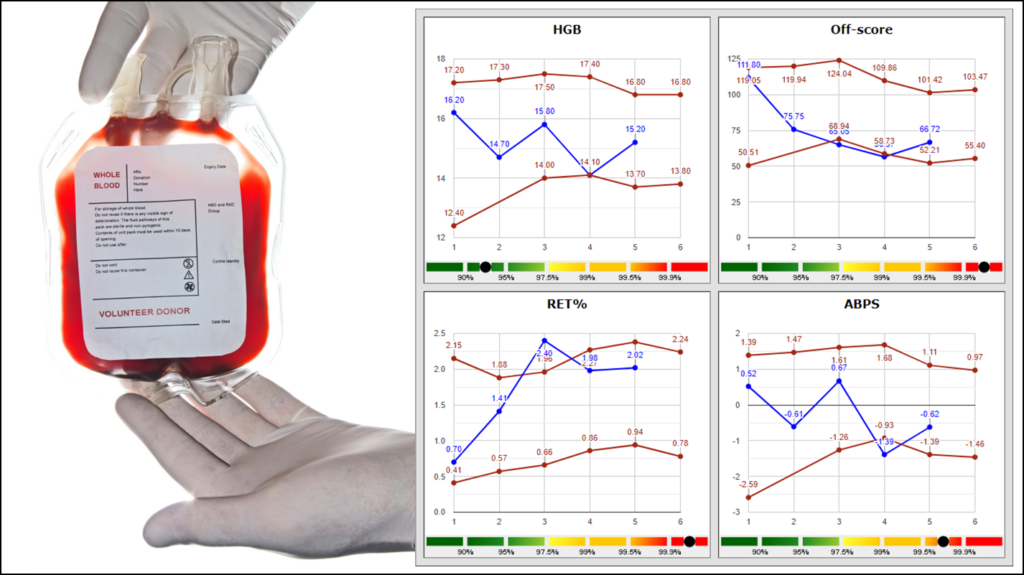
For the effectiveness of the ABP, a frequent sample collection both in- and out-of-competition is of utmost importance 51and standardized sample collection and analysis protocols as well as internal and external quality control systems need to be employed to reduce the pre-analytical and analytical variability to a minimum.52 As indicated above (Section 3), the required EDTA whole blood samples have to be transported refrigerated (4-15°C) and temperature-controlled.53 The maximum allowed transport time depends on the temperature and varies between 60 and 35 hours. Following sample reception at the anti-doping laboratory, blood specimens usually have to be analyzed within 12 hours by using a harmonized protocol and a defined hematological analyzer. Before, during, and after sample analysis, three-level internal quality controls are measured to ensure stable and precise analytical conditions. Following homogenization for at least 15 min at room temperature, each sample is measured twice and results are only considered valid if the absolute differences between the values for Hb and Ret% are within an acceptable range. Test results are anonymously submitted into ADAMS, which allows matching of the anonymous results obtained by different laboratories with the individual longitudinal hematological profile of an athlete.54 The adaptive model in ADAMs automatically evaluates the sample and updates the profile, before data are anonymously reviewed by an ABP Management Unit (APMU), which is usually hosted by an anti-doping laboratory and responsible for the timely passport management. Atypical profiles as exemplary displayed in Figure 3 are further evaluated by internal and/or external experts in hematology, sports medicine, and exercise physiology to decide whether doping or other physiological/pathological reasons caused the results.
3.1.1.2 Other detection strategies
Besides the hematological module of the ABP, also other indirect strategies to provide evidence for autologous blood doping have been discussed in the past. The quantification of urinary plasticizers and their metabolites by using liquid chromatography-tandem mass spectrometry (LC-MS/MS) is considered a promising approach as phthalates such as di-(2-ethylhexyl)phthalate (DEHP) are usually added to the polyvinyl chloride (PVC) used for the production of blood bags to improve their flexibility.55 During ex vivo storage, they diffuse into the stored blood and elevated urinary levels of phthalate metabolites were observed for approximately 48 hours in subjects having received blood transfusions.56 However, as plasticizers are also released from numerous products made of PVC such as food packaging and toys, they can be found in almost every human being, and urine levels are subject to a considerable intra- and inter-individual variability.57 Therefore, abnormally high concentrations should not be used as sole evidence for an anti-doping rule violation, but rather support the interpretation of atypical hematological ABP profiles.
As mentioned above, the refrigerated ex vivo storage of RBCs results in the development of storage lesions, which include, inter alia, membrane losses by vesiculation, protein and lipid oxidation, the formation of echinocytes, a reduced cellular flexibility, and declines of the intracellular pH and adenosine triphosphate (ATP) and 2,3-diphosphoglycerate (2,3-DPG) levels.58 While some changes are reversible, others permanently affect the post-transfusion functionality and viability of the cells. Especially irreversible storage lesions could represent promising biomarkers for the reinfusion of stored blood and thus the misuse of autologous blood transfusion.59 Two studies have investigated the storage-induced changes of the cytosolic60 and membrane61 proteome of RBCs, and identified several candidates which might be employed for blood doping detection in the future. However, their detection in the blood of doping athletes following reinfusion could be challenging as the stored cells will only constitute a few percent of total circulating RBCs and especially the cells with severe storage lesions are eliminated within the first 24 hours.62 A third study published without peer-review investigated the cytosolic RBC peptidome of healthy subjects before and following autologous blood transfusion with cryopreserved RBCs and the authors could establish a valid discriminating multivariate model for the detection of autologous blood doping.63
Another promising approach for the detection of autologous blood doping is the measurement of total Hb.64 Compared to the blood Hb concentration monitored within the hematological module of the ABP, the total Hb mass doesn’t depend on fluctuations of plasma volume.65 Currently, it can only be determined by carbon monoxide (CO) rebreathing where a spirometer is used for rebreathing a known volume of CO over a period of 2 min.66 Both before and after the intervention, the concentration of carboxyhemoglobin (CO-Hb) is measured in capillary blood samples obtained from the ear lobe. Then, the total Hb mass can be calculated on the basis of the difference in CO-Hb before and after CO application, the volume of CO, and the binding capacity of Hb for CO. But until an alternative to CO rebreathing is available, the practical applicability of this method to sports drug testing is limited as CO is toxic, can temporarily reduce athletic performance, and the athlete has to fully cooperate with the doping control officer to obtain reliable results.67
Since the introduction of the hematological module of the ABP in 2009, more than 150 athletes have been directly sanctioned on the basis of ABP data.68 Additionally, several athletes were banned from competing as their blood bags and equipment for blood withdrawal, storage and transfusion were confiscated during raids at team quarters and the residences of team physicians.69 Both these data and incidents demonstrate that blood doping is still misused by athletes and points to the need for continuous improvement in detection strategies.
3.1.2 Homologous blood transfusions
The use of autologous blood transfusions either requires careful timing of blood withdrawal and re-infusion, especially before important competitions,70 or is associated with high efforts and costs when cryo-conservation is employed to prolong the otherwise limited storage time.71 Therefore, some athletes might consider homologous blood doping as a valuable alternative doping strategy, even though this approach for performance-enhancement is more readily detected. Here, RBC concentrates are obtained from a donor with matching AB0 and D blood groups to avoid life-threatening hemolytic reactions.72 As the blood can be directly transfused following collection, the negative effects of interim storage are avoided.73 But unless an identical twin is available, some minor blood group antigens will inevitably be mismatched and can therefore be employed as analytical targets in sports drug testing. So far, 29 different blood group systems with a total of 245 antigens have been described,74 and every RBC of one individuum has the same, unique combination of antigens on its surface, whose composition is genetically determined.75 Consequently, the presence of mixed RBC populations is a strong indication for the misuse of homologous blood transfusions, as the only other – rather unlikely – explanations for this phenomenon are a preceding hematopoietic stem cell transplantation or blood group chimerism, a very rare event (< 1 case in 1 million) which can easily be proven by DNA analysis.76
For the differentiation of mixed RBC populations, both high and low incidence blood groups are not very useful.77 But antigens found with a moderate frequency such as the Duffy/FY (FYa, FYb) and Kidd/JK (Jka, Jkb) are very informative and were therefore employed for the development of doping control detection assays. At first, different serological tests based on hemagglutination were utilized:78 In the presence of a specific antiserum, mixed RBC populations lead to an incomplete agglutination (“mixed field reaction”), which can either be detected by microscopic evaluation or separation of the sample on a special gel containing the agglutinating antibodies.79 While antigen-negative cells can just pass through the gel, antigen-positive cells agglutinate and remain on top of it. This approach can sensitively detect even small amounts (down to 5%) of antigen-positive cells in a population of antigen-negative cells, however, it can be difficult to recognize a minority of cells missing an antigen among agglutinated cells.80 In 2003, a new test for the quantitative determination of antigenic RBC profiles by using flow cytometry was introduced, which can also detect small populations of antigenically distinct cells (<5%), irrespective of whether or not they carry a certain surface marker.81 Blood group-specific antibodies and fluorochrome-conjugated secondary antibodies are employed to label cells with the respective surface marker before passing a beam of excitation light (e.g. an argon ion laser operating at a wavelength of 488 nm) in a fluid stream.82 Each cell is analyzed separately and different detectors measure the emitted/scattered light, which gives information about cell size/volume (forward scatter FSC), intracellular structures (side scatter SSC), and – if a fluorescent dye is attached via an antigen-specific antibody – the presence/absence of surface structures. Labeled antigen-positive and unlabeled antigen-negative RBCs are finally evaluated by using a single-parameter histogram. The initial setup of this approach comprised twelve blood group antigens: S, s (MNS system), C, c, E, e (RH system), K, k (KEL system), FYa, FYb (FY system), and Jka, Jkb (JK system).83 However, optimized panels include only eight of these markers.84
The infusion of an RBC concentrate with a volume of ~ 450 mL will dilute the recipient’s blood (volume: ca. 5000 mL) by approximately 10%,85 however, about 25% of the transfused cells are eliminated from the circulation of the recipient within 24 h as a result of cellular ageing and accumulated storage lesions.86 But as RBCs have a total lifespan of approximately 120 days, at least some donor cells should be present for several weeks and therefore allow for a sensitive detection of homologous blood doping.87
During the 2004 Olympic Games in Athens, Greece, the flow cytometric detection assay for homologous blood transfusions was used for the first time and cyclist Tyler Hamilton was tested positive after winning the gold medal in the men’s individual time trial.88 But as his B sample was mistakenly stored frozen, he could only be sanctioned after a blood sample collected a few weeks later during the Vuelta a Espana also yielded positive results for homologous blood doping. After initially claiming to be a natural blood group chimera,89 he finally confessed autologous blood doping,90 and a mix-up of RBC concentrates was assumed as explanation for the AAFs obtained in 2004.91 So far, only 10 AAFs with homologous blood doping were reported by the WADA.92 It can be assumed that cheating athletes either switched to doping practices more difficult to detect or use donors with largely identical antigenic profiles for homologous blood doping.93 Potential strategies to reduce the risk for such false-negative samples would be to use a larger panel of antigens or DNA analysis.94 But as nucleated donor white blood cells are required for the identification of foreign DNA in the recipient’s blood, the transfusion of leuko-depleted RBC concentrates can limit the effectiveness of such approaches.
3.2 Blood as biological matrix in sports drug testing
3.2.1 Human growth hormone (hGH)
hGH is a peptide hormone secreted by the pituitary gland in a pulsatile manner, and comprises different variants resulting from alternative splicing and post-translational modifications such as fragmentation and phosphorylation.95 The main endogenous isoform accounting for more than 70% of circulating hGH consists of 191 amino acids and has a molecular mass of 22 kDa. It mediates its growth promoting and metabolic functions through binding to the GH receptor present on every cell of the human body, which either directly affects biological processes such as carbohydrate and lipid metabolism, or stimulates the synthesis of insulin-like growth-factor I (IGF-I) in the liver, a peptide responsible for most anabolic effects of hGH.96
The main therapeutic indication for hGH administration is the treatment of GH deficiency in children and adults.97 While extracts from human cadaver pituitary glands were initially used as a pharmaceutical source for hGH, it has also been recombinantly produced since 1985. Recombinant hGH (rhGH) has the same amino acid sequence as the pituitary 22 kDa isoform and can neither be distinguished from it by analyzing the isotopic ratio of 13C/12C with carbon isotope ratio mass spectrometry (IRMS), nor on the basis of species/cell line-specific differences in post-translational modifications as EPO (see also 3.2.2).98 rhGH and pituitary extracts are also distributed on the black-market and therefore readily available for doping athletes. The misuse of such preparations is associated with unforeseeable health risks as black-market products are often incorrectly labeled – regarding both their qualitative and quantitative composition.99 Moreover, the administration of rhGH to healthy individuals – especially at supratherapeutic doses – can lead to severe adverse effects similar to the symptoms of acromegaly caused by a pathological increase in endogenous GH production including musculoskeletal (e.g. prognathism), metabolic (e.g. diabetes mellitus), and cardiovascular changes (e.g. hypertension, cardiomyopathy).100 The use of pituitary extracts from human cadavers additionally poses a risk of infection with Creutzfeld–Jakob disease.
Even though no studies exist which clearly confirm the performance-enhancing effects of GH in healthy adults, anecdotal evidence suggest that the peptide hormone is frequently misused by cheating athletes because of its presumed lipolytic and anabolic actions, especially when combined with anabolic steroids and insulin.101 hGH is often detected in material confiscated by police and customs102, and several athletes have confirmed the misuse of this drug.103 Moreover, Victor Conte, the central figure of the so called “BALCO scandal” uncovered in 2003, claimed that he treated many elite athletes including Marion Jones with hGH.104 Consequently, the misuse of hGH (and its mediator IGF-I) in sports has been prohibited at all times since 1989 and the peptide is included in section S2 (Peptide Hormones, Growth Factors, Related Substances and Mimetics) of the WADA Prohibited List.105
The detection of hGH misuse in sports is very challenging as recombinantly produced GH cannot be distinguished from the major endogenous isoform.106 Moreover, the pulsatile secretion of the hormone leads to highly variable plasma concentrations (0.1-18 ng/mL) and its release from the pituitary gland has been shown to be stimulated by sleep, stress, and physical exercise.107 At the same time, the peptide hormone has a very short biological half-life of 15-20 min 108 and only 0.01-0.001% is renally excreted, resulting in urinary concentrations of 4 pg/mL or even lower.109 Furthermore, urinary elimination is not necessarily a constant function of GH blood levels as both the glomerular filtration and tubular reabsorption of the peptide hormone can be affected by pathological conditions such as diabetes mellitus as well as strenuous physical exercise.110 Consequently, blood is the preferred biological matrix for the detection of GH.
In principle, two different analytical strategies have been employed to detect hGH misuse in sports: The first, direct approach measures hGH isoforms from serum by using two different sandwich-type chemiluminescence immunoassays.111 While the first kit recognizes all pituitary isoforms of hGH, the other one is highly specific for the monomeric 22 kDa isoform corresponding also to rhGH. As the exogenous administration of rhGH results in a decreased release of pituitary hGH due to negative feedback mechanisms mediated by IGF-I, the 22 kDa isoform becomes predominant and the ratio relative to the other variants changes.112 By analyzing a doping control sample with both assays, the relative abundance of the 22 kDa isoform can be calculated as rec/pit ratio and subsequently be employed to provide evidence for the misuse of rhGH. To be compliant with WADA guidelines,113 two different sets of immunoassays based on antibodies specific for different epitopes were developed for screening and confirmation purposes (pit A/B and rec A/B).114 For each kit, individual gender-specific thresholds have been defined by evaluating numerous athlete’s samples (Kit 1: males – 1.84, females – 1.63; Kit 2: males – 1.91, females – 1.59),115 and if the decision limit is exceeded during initial analysis, the respective sample has to be re-analyzed with both available kits. Additionally, the serum concentration of rhGH is determined and if the value is below 150 pg/mL, samples are reported negative irrespective of the rec/pic ratio. Due to the short biological half-life of hGH, the detection window of the isoform approach is limited to 24-36 h post-administration.116 As it is therefore rather unlikely to catch athletes misusing rhGH by in-competition testing, frequent out-of-competition testing is of utmost importance.117 Moreover, this approach cannot detect the misuse of cadaveric hGH, IGF-I, or agents stimulating endogenous hGH production.
By contrast, the “biomarker approach” targets two hGH-dependent markers whose serum concentrations were found to be significantly elevated following rhGH administration: IGF-I and the N-terminal extension peptide of procollagen type III (P-III-NP).118 These analytical targets were identified within the large multi-center GH-2000 project from an initial set of 25 potential markers and are characterized by a longer biological half-life and more stable serum levels than hGH.119 But as they are naturally present in the human body, only excess levels which do not occur under any physiological or pathological condition can be employed to provide evidence for hGH misuse.120 Comprehensive data obtained from an administration study was employed to construct gender-specific formulas for the identification of subjects treated with rhGH.121 These discriminant function formulas combine the results obtained for both biomarkers with a correction factor for the athlete’s age as both IGF-I and P-III-NP levels were found to decline with increasing age. The resulting GH-2000 score is compared to the corresponding decision limit defined for the respective gender and used assays.122 Both the formulas and decision levels were further optimized over the years when the samples of several thousand male and female athletes were evaluated.123 According to current WADA guidelines,124 a two-site solid-phase immunoradiometric assay (IRMA), an automated sandwich, chemiluminescent immunoassay (CLIA), or a liquid chromatography (high-resolution) (tandem) mass spectrometry (LC-(HR)MS(/MS)) method can be employed for the quantification of serum IGF-I. In all three approaches, serum complexes between IGF-I and different binding proteins first need to be dissolved by sample acidification and IGF-II is then added in excess to prevent re-aggregation. By contrast, P-III-NP levels can be determined by using either a competitive radioimmunoassay or an automated two-site sandwich chemiluminescent immunoassay. An alternative mass spectrometric assay is under development.125 In case of a presumed AAF, two different combinations of the abovementioned assays have to be used for confirmation purposes.126 For all measurements, serum samples are required as the urinary levels of IGF-I are significantly lower than in blood and do not necessarily correlate with serum concentrations.127 The evaluation of administration study data indicates detection windows up to two weeks following cessation of rhGH administration, but they will strongly depend on the applied dose and gender of the athlete as men were found to show a stronger IGF-I response than women.128
Since the implementation of the isoform assay in 2004 and the biomarker approach in 2012,129 a total of 38 AAFs with hGH were reported by the WADA.130 The first AAF was detected in November 2009 when the isoform approach returned positive for an out-of-competition sample of the British rugby player Terry Newton.131 Since then, most of the positive samples were identified by using the isoform assay, however, the biomarker approach yielded also two AAFs for powerlifters misusing hGH during the London Paralympic Games in 2012.132
3.2.2 Erythropoietin-receptor agonists (ERAs)
In humans and many other species, the production of RBCs is regulated by EPO, a highly glycosylated peptide hormone produced mainly by the kidneys.133 EPO comprises 165 amino acids and has a molecular mass of approximately 30 kDa, of which ~ 40% can be attributed to carbohydrate moieties attached via three glycosylation sites. As these glycan structures vary slightly in their complexity, sulfatation, branching degree and sialic acid content, EPO comprises a series of isoforms.134
Since the late 1980s, EPO can be recombinantly produced in Chinese hamster ovary (CHO) cells and has clinical approval for the treatment of anemia in patients with renal failure.135 The first recombinant EPO (rEPO) preparations are Epoetin α and β (“biosimilars”), which can only be distinguished from the endogenous protein by their glycosylation/sulfatation patterns as the cell line used for their production has a slightly different enzymatic equipment. In the following years, plenty other EPO biosimilars were developed by using CHO, baby hamster kidney (BHK) and human cells, including also genetically and chemically modified variants with a prolonged biological half-life (“biobetters”). While in the novel erythropoiesis-stimulating protein (NESP; Aranesp®), five amino acids of the original sequence were replaced to introduce two additional carbohydrate chains,136 the continuous EPO receptor activator (CERA; Mircera®) bears a polyethylene glycol (PEG) residue.137
Due to the significant performance-enhancing effects as well as the obvious advantages over the use of blood transfusions regarding accessibility and handling, rEPO and its derivatives quickly aroused the interest of cheating athletes, especially in endurance disciplines.138 But even though the administration of EPO is considered safer than the use of blood transfusion, the increased blood viscosity can cause several adverse effects such as thrombosis.
Although the misuse of EPO was directly prohibited by the IOC in 1990, it took 10 years before the first direct doping control assay was introduced, as a very high sensitivity and specificity are required to detect the very low urinary concentrations of the drug and distinguish synthetic from endogenous EPO.139 LC-HRMS/MS is currently no option as endogenous human EPO and first generation Epoetins differ only in the composition of a few sugar moieties.140 But as mentioned above, the production of EPO by different cell lines results in a varying post-translational modification (glycosylation/sulfatation) and thus differential isoform profiles, which can be detected by isoelectric focusing (IEF) and double-Western blotting of urinary retentates obtained by ultrafiltration.141 During IEF, proteins are placed in a pH gradient and separated according to their isoelectric point (pI), which depends on their amino acid composition as well as the presence of post-translational modifications.142 In most cases, this technique was found to work well for urine specimens, but occasional changes of urinary IEF-profiles due to enzymatic activity (“active urines”) and exercise (“effort urines”) necessitated the development of a complementary detection strategy based on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).143 Here, EPO variants are separated by their molecular mass, which differs between endogenous human EPO and the different biosimilars. Moreover, this approach was found to be beneficial regarding the detection of Epoetin δ/Dynepo®, which is recombinantly produced in a human cell line and whose IEF profile is therefore more difficult to distinguish from endogenous EPO.144
With the development of the PEGylated biosimilar CERA, the use of urine for EPO detection became insufficient as both the long biological half-life (~130 h) and high molecular mass (~ 60 kDa) of the protein conjugate result in a very limited renal excretion.145 Even though an exercise-induced proteinuria can occasionally result in CERA urine levels detectable by conventional IEF analysis,146 both the IEF- or SDS-PAGE-based approaches had to be modified to allow for a detection of different EPO variants from plasma/serum.147 Moreover, the use of plasma/serum is generally beneficial as serum EPO is – in contrast to urinary EPO – neither affected by exercise, nor subject to enzymatic degradation.148 But due to the high protein (60-80 g/L) and at the same time low EPO content (30-170 ng/L) of plasma/serum, an additional immunopurification 149 or immunodepletion step 150 is required before samples can be further analyzed. And especially the SDS-PAGE approach had to be largely altered as the PEG moiety of CERA is bound by the detergent SDS, which significantly reduces the assay’s sensitivity due to a decreased interaction with the primary antibody used during Western blotting.151 However, the use of sarcosyl (SAR) instead of SDS as detergent in the sample and running buffer could solve the problem as it only binds to the protein part of the molecule. When compared to CERA post-administration serum samples analyzed by IEF, SAR-PAGE was found to be at least 6 times more sensitive and could prolong the detection window by 7 days.152
Alternatively, also an ELISA employing an anti-EPO capturing and anti-PEG detection antibody can be used to test serum samples for CERA,153 but as this approach cannot simultaneously detect other EPO biosimilars, its use always necessitates further analyses to cover all analytes.154 According to current WADA guidelines,155 a preceding immunopurification is required both for urine and plasma/serum analysis to avoid effects of protein overloading such as band deformation/smearing,156 and all three abovementioned approaches can be employed as initial testing procedure.157 For confirmation purposes, SAR- or SDS-PAGE have to be used for the detection or rEPO and IEF, SAR- or SDS-PAGE for genetically or chemically modified EPO biosimilars. An additional challenge represents the rare EPO variant c.577del, which predominantly occurs in East Asian populations (frequency of 0.5-1%) and has a 27 amino acid extension resulting in a similar molecular mass and electrophoretic behavior as rEPO.158 When analyzing serum/plasma samples by SAR-/SDS-PAGE, the bands corresponding to c.577del EPO and rEPO can be clearly distinguished by their characteristic shape: While the EPO c.577del variant and WT-EPO show a well-defined double-band pattern, rEPO is a smear above WT-EPO and usually both bands are not completely resolved. In urine, the differentiation of c.577del EPO and rEPO is much more difficult as the characteristic double band is not always present. Therefore, further investigations such as the evaluation of additional blood samples are required in cases of AAFs with rEPO in urine. This is another example why the use of plasma/serum for ESA analysis is superior to urine.
In addition to these direct detection methods, also an indirect approach can be employed to provide evidence for EPO misuse. As indicated above, this method is based on the measurement of different blood parameters with a hematological analyzer/specific immunoassays, and their evaluation by using two different discriminant models.159 The ON-model originally combined 5 variables (hematocrit (Hct), reticulocyte hematocrit (RetHct), percent macrocytes (%Macro), serum EPO (EPO), and soluble transferrin receptor (sTfr)) to identify current EPO misuse. By contrast, the OFF-model for the detection of recent EPO misuse evaluated 3 parameters as markers of an altered erythropoiesis (Hct, RetHct, and EPO). By comparing the resulting scores to a population-derived threshold, athletes that are/were doping with EPO could be successfully identified. In the following years, both models were further optimized160 and the OFF-hr score based on the Hb and Ret% 161 is currently evaluated within the hematological module of the WADA ABP,162 where the results are compared to subject-specific hematologic reference values instead of population-derived thresholds.163 Furthermore, the ABPS is calculated, which is not only sensitive to EPO misuse but also other blood doping practices.164 As the effects of EPO on the hematological profile of an athlete can last longer than the presence of the drug in circulation, such an indirect doping control assay based on the longitudinal monitoring of multiple biomarkers can enable a more sensitive and prolonged detection of EPO misuse and identify subjects for targeted testing.165
Besides the abovementioned EPO biosimilars, also other ESAs and EPO-receptor agonists (ERAs) have been developed over the years. Several drug candidates are therapeutic fusion proteins or protein conjugates with a high molecular mass, which results in a prolonged biological half-life and reduced urinary excretion. Therefore, plasma or serum are the preferred biological matrix for sports drug testing purposes. Peginesatide (HematideTM) is a PEGylated homodimeric EPO-mimetic peptide with a MW of approximately 45 kDa, which has no structural resemblance to EPO but can nevertheless bind to and activate the EPO receptor.166 For a short period of time, the drug had clinical approval for anemia therapy in patients with chronic kidney disease, but it was withdrawn from the market due to several cases of serious hypersensitivity reactions.167 Epo-Fc (Fc = fragment crystallizable) represents a recombinant fusion protein of one or two EPO molecules and the dimeric Fc domain of human immunoglobulin G (IgG), which was designed to allow for a pulmonary administration via the neonatal Fc receptor.168 Sotatercept (ActRIIA-Fc) and Luspatercept (modified ActRIIB-Fc) are dimeric fusion proteins composed of the extracellular domain of the human activin receptor type IIA or B (ActRIIA/B) and the Fc fragment of human IgG, which act as ligand traps for different cytokines of the transforming growth factor beta superfamily (TGF-β) involved in the negative regulation of RBC differentiation and maturation.169 Due to the resulting erythropoiesis-stimulating effect independent from the EPO receptor, both protein drugs are promising therapeutics for the treatment of anemia in rare blood diseases and Luspatercept has gained clinical approval in 2019/2020. EPO-Fc, Sotatercept and Luspatercept could be successfully implemented into existing multi-analyte IEF, SDS- and SAR-PAGE approaches.170 Moreover, complementary methods based on ammonium sulfate precipitation, (immuno-)affinity purification, tryptic digestion, and LC-MS/MS were developed, which allow for an unambiguous identification of the target analytes at the amino acid sequence level.171 By contrast, a separate method was developed for the detection of Peginesatide from plasma, which employs protein precipitation, subtilisin digestion, and LC-MS/MS.172
Between 2003 and 2020, a total of 953 AAFs with EPO have been reported via ADAMs173 and many of these findings were a direct result of targeted testing due to atypical hematological profiles observed within the ABP.174
3.2.3 Hemoglobin-based oxygen carriers (HBOCs)
As mentioned above, the oxygen transport capacity of the blood is a limiting factor in endurance sport disciplines.175 Besides blood transfusions (→ 3.1) and ESAs (→ 3.2.2), also artificial oxygen carriers can be misused by athletes for performance enhancement. These compounds are based on human, bovine or recombinant Hb, and were/are developed as substitutes for blood transfusions. Hb is the main erythrocytic protein and in many species responsible for the transport of oxygen from the lungs to the tissues.176 Each of its four protein subunits (2 x α, 2 x β) contains a prosthetic heme group with a central iron ion (Fe2+), which can bind one molecule of O2. The oxygen affinity of the molecule depends on the surrounding pH and carbon dioxide concentration and is additionally modified through the interaction with modulators such as 2,3-DPG. Unfortunately, it is not possible to simply use purified human Hb as blood substitute, as free Hb not encapsulated by RBCs is rapidly degraded.177 The glomerular filtration of the resulting αβ-dimers leads to kidney damage as they precipitate in the renal tubules. Moreover, scavenging of nitric oxide (NO) by free Hb causes vasoconstriction, because NO is an important regulator of smooth muscle relaxation in blood vessels. And as the concentration of 2,3-DPG is significantly lower in blood than in RBCs, the efficiency of free Hb to bind and release oxygen is considerably reduced, making it rather an oxygen reservoir than oxygen transporter.
Consequently, different strategies for hemoglobin stabilization including polymerization (e.g. with glutaraldehyde or raffinose), intra-molecular cross-linking (e.g. with dibromosalicyl fumarate (DBBF), conjugation (e.g. with PEG), and encapsulation were employed during HBOCs development.178 However, most products still were found to cause severe side effects which led to an early termination of clinical development. So far, only one product has at least regionally (South Africa and Russia) gained clinical approval for the treatment of acute anemia in humans: HemoPure® (HBOC-201/Hemoglobin glutamer-250 [bovine], Biopure, Cambridge, MA)) which is composed of purified bovine Hb polymerized with glutaraldehyde.179 Moreover, a structurally related product called Oxyglobin® (Hemoglobin glutamer-200 [bovine]) is approved for veterinary use in dogs both in Europe and the US. According to the manufacturer,180 more than 240,000 units of Oxyglobin® have been sold since its clinical approval in 1999 and approximately 30 species were treated off label. A few other promising HBOCs are still in clinical development:181 Sanguinate (Prolong Pharmaceuticals, South Plainfield, NJ) is based on PEGylated bovine carboxyhemoglobin and currently undergoing Phase I/II clinical trials.182 While the attachment of the PEG moiety increases the biological half-life and modifies the oxygen affinity of the molecule, the release of carbon monoxide reduces vasoconstriction, inflammation, and apoptosis. By contrast, the company Hemarina (Morlaix, France) has developed different products (HEMOXYcarrier® and HEMO2life®) containing extracellular Hb from the lugworm Arenicola marina as active ingredient.183 The structure of this molecule differs substantially from human Hb as it has a molecular mass of 3600 kDa (instead of 64 kDa) and can simultaneously bind 156 oxygen molecules. Due to this enormous size, it does not scavenge NO in the endothelial intercellular space. The products based on this lugworm Hb can not only be used as blood substitutes but also as additives to organ preservation solutions for transplantations and are currently investigated in Phase 2/3 clinical studies.184
Due to the presumed performance-enhancing effects, HBOCs were added to the IOC’s list of prohibited substances in 2000.185 Even though the short biological half-life of 13-72 h and the immediate effect following intravenous administration strongly suggest a misuse in-competition only,186 they are currently prohibited at all times.187 Compared to blood transfusions, the use of such HBOCs would be beneficial regarding the logistic requirements and the shelf life/storage time of the doping agent.188 And in contrast to homologous blood doping, no blood group cross-matching is required and the risk for pathogen transmission significantly reduced. Even though no AAFs with HBOCs have been reported so far, 189 several cyclists have confirmed to have misused Oxyglobin® in the past.190 To have a measure against HBOCs doping, different detection strategies were developed by anti-doping laboratories. All these approaches use blood as biological matrix as the high molecular mass of most HBOCs results in only little or no urinary excretion.191
A very simple initial screening for HBOCs misuse can be the visual evaluation of blood samples following centrifugation, which is based on the fact that the appearance of blood changes in the presence of free hemoglobin (Figure 4).192 It could be demonstrated that such an approach can detect less than 2 g/L of Hemopure® in plasma, which would result in theoretical detection windows of at least 3 days (considering a biological half-life of 24 h and a dose of 250 mL, leading to an initial plasma Hb concentration of 13 g/L). An alternative initial testing procedure determines the circulating amount of free Hb by measuring total and erythrocytic Hb with a hematological analyzer, and a cut-off of 3.5 g/L was suggested to differentiate negative from suspicious samples.193 Another approach employs native-gel electrophoresis and Western blotting for the differentiation of different HBOCs from endogenous Hb released by hemolysis in serum immunodepleted from the protein haptoglobin, which has a strong affinity for free Hb.194
If one of these screening assays yields suspicious results for a plasma/serum sample, confirmation methods need to be employed to distinguish RBC hemolysis (e.g. due to sample handling or pathological conditions) from HBOCs misuse. Potential strategies comprise size-exclusion chromatography (SEC) in combination with UV/VIS spectrophotometry to differentiate intact cross-linked/polymerized HBOCs from dissociated endogenous Hb,195 LC-MS/MSto detect tryptic peptides derived from bovine Hb,196 and capillary electrophoresis (CE) either in combination with UV/VIS for the detection of molecules separated by their electrophoretic mobility197 or MS to additionally distinguish bovine and human Hb monomers by their molecular mass following in-source protein dissociation.198 Depending on the analyte and the sample volume, the sensitivity of these assays varies between 0.2 g/L199 and 5.5 g/L.200 As different studies suggest that the Hb concentration has to increase by at least 5% to have any performance-enhancing effects, the minimum detection level required for sports drug testing procedures is 8 g/L.201 Currently, all of the above-mentioned approaches can be employed for the qualitative detection of HBOCs from doping control blood/plasma/serum samples as long as the requirements of the WADA’s International Standard for Laboratories (ISL) for screening and confirmation are fulfilled.202
3.2.4 Insulin an IGF-I analogues
Insulin is a small peptide hormone produced by the β-cells of the pancreas and the key regulator of carbohydrate metabolism.203 It consists of and A- and B-chain (21 and 30 amino acids) connected by two disulfide bonds and has a molecular mass of approximately 5.8 kDa. Following glucose-induced synthesis and secretion, insulin stimulates the uptake of glucose into muscle and adipose tissue as well as its storage as glycogen. Moreover, glycogen breakdown and gluconeogenesis in the liver are inhibited. Similarly, also the uptake of amino acids and fatty acids and their storage in the muscle/fat tissue are stimulated and proteolysis and lipolysis inhibited.
In 1982, recombinantly produced human insulin was introduced to the market and a “game-changer” in the therapy of Diabetes mellitus, which represents one of the biggest health issues of the 21st century.204 Recombinant human insulin is usually administered by subcutaneous injection and as the monomers of the peptide have a strong tendency for self-association, especially when zinc ions are present, it takes a lag-phase of 45-120 min until the resulting hexamers dissociate into bioavailable monomers.205 To improve the pharmacokinetic properties and especially controllability of the drug, different synthetic analogs have been developed which have either rapid effects on blood sugar levels or ensure constant basal insulin plasma levels:206 Rapid-acting insulins such as Humalog (insulin lispro), Novolog (insulin aspart), and Glulisine (insulin apidra) are slightly modified to reduce their tendency for self-association and thus increase the bioavailability to 10-15 min following subcutaneous injection.207 By contrast, the structural changes of slow-acting insulins as for example Lantus (insulin glargine), Tresiba (insulin degludec), and Levemir (insulin detemir) result either in a delayed absorption from the injection site due to the formation of micro-precipitates (Lantus) or an increased self-association and binding to serum albumin (Tresiba, Levemir).208
Due to their anabolic and anti-catabolic properties, insulin and its synthetic analogs have also the potential for being misused as performance-enhancing agents in sports.209 Therefore, they were added to the WADA prohibited list in 1999210 and different strategies for their detection from doping control samples have been developed.211 Even though insulins are renally eliminated, metabolic degradation results in very low urine concentrations of less than 0.1 ng/mL.212 Therefore, several concentration and purification steps as well as a highly sensitive instrumentation are required to enable their urinary detection. Routinely, this can be successfully accomplished by subjecting urine samples to solid-phase extraction (SPE) or ultrafiltration and immunoaffinity purification with magnetic beads and specific anti-insulin antibodies, before LC-(HR)MS/MS is employed to distinguish endogenous human insulin from the synthetic derivatives, which differ only slightly in their amino acid sequences.213 The resulting detection limits are as low as 5 pg/mL.
However, the use of plasma/serum instead of urine for insulin detection can be beneficial for several reasons: Plasma/serum levels can be significantly higher (up to 3 ng/mL after glucose-induced stimulation214 and knowledge on the metabolic fate of the analytes is not required as the intact insulins can be detected.215 This is of particular importance for the long-acting insulins Lantus (insulin glargine) and Levemir (insulin detemir)216: Due to the prolonged serum half-life, both protein drugs are extensively metabolized and not excreted into urine in their intact form (except for Tresiba). But while different unique degradation products such as desB30-32 have been described for Lantus, the main urinary metabolite desB30 of Detemir cannot be distinguished from endogenous human insulin. Therefore, plasma/serum is currently the only matrix that can be employed to test for doping with Detemir. But the detection of insulins from blood is also associated with some analytical challenges as the basal concentrations of endogenous insulin are still low (0.2-0.5 ng/mL), the sample volume available for doping control analysis is limited (ca. 50-250 µL), and highly abundant plasma proteins such as albumin are present in the samples.217 Moreover, hemolyzed samples can be problematic due to significant matrix effects and insulin degradation in the presence of hemoglobin.218 By using modified versions of the abovementioned protocols comprising sample dilution, ultrafiltration or protein precipitation, immunoaffinity purification and LC-HRMS/MS, a highly sensitive and specific detection of insulin and its derivatives down to concentrations of 0.1 ng/mL (0.8 ng/mL for detemir) can be accomplished in non-hemolyzed plasma/serum samples.219 Alternatively, also protein precipitation and mixed-mode cation-exchange SPE can be employed prior to LC-HRMS/MS analysis.220
As mentioned above (3.2.1), IGF-I is a peptide synthesized by the liver which mediates many anabolic effects of hGH. It comprises 70 amino acids, has a molecular mass of 7.6 kDa, and three intramolecular disulfide bonds.221 In vivo, most of the circulating IGF-I is bound by special binding proteins which protect the peptide from proteolytic degradation and prolong its biological half-life. Due to its growth promoting activity, IGF-I is a therapeutic agent for the treatment of growth disturbances due to IGF-I deficiency, and different synthetic analogs with a reduced affinity for endogenous binding proteins have been developed to improve the pharmacokinetic properties of the peptide.222 These derivatives comprise R3-IGF-I, where the glutamic acid (E) at position three of the amino acid sequence is replaced with an arginine (R) residue, LongR3-IGF-I with an additional N-terminal extension of 13 amino acids, and Des1-3-IGF-I, a truncated derivative lacking the first three amino acids at the N-terminus.223 Even though no drug candidate except for recombinant human IGF-I (Mecasermin, brand name Increlex) has gained clinical approval yet, all preparations are readily available for cheating athletes as they are distributed for research purposes and on the black market.224Analogous to hGH, the misuse of IGF-I and its analogs in sports is prohibited at all times225 and different doping control detection assays employing immunoaffinity purification (alone or in combination with preceding SPE or protein precipitation) and LC-MS/MS for the analysis of urine or plasma/serum samples were established.226 In theory, all analytes can be sensitively detected from both biological matrices, however, data on the in vivo elimination of the different IGF-I analogues in humans is unfortunately not available as they are unapproved substances.227 As it remains to be elucidated if the parent compounds or only some metabolites/degradation products are renally eliminated, plasma/serum might currently be the more useful biological matrix for the detection of IGF-I and its analogues, especially since stability studies showed that LongR3-IGF-I is not stable in urine and only the degradation product Des1-10-LongR3 -IGF-I can be detected.228 A very recent approach employing so-called IRIS (isotope-labeled reporter ion screening) allowed for determining degradation products of hGH and IGF-I through skin-borne proteases in human plasma.229 How and to which extend these degradation products can contribute to enhancing anti-doping efforts concerning IGF-I and hGH remains to be shown in future studies.
Since the addition of insulin and its derivatives to the WADA Prohibited List, a total of 17 AAFs have been reported and most of them can presumably be attributed to diabetic athletes.230 For synthetic IGF-I analogues, no positive findings were obtained so far, maybe because almost exclusively urine specimens were analyzed in the past.
3.2.5 Steroid esters
Since decades, AAS including testosterone and its derivatives represent the substance class most frequently misused in sports, even though severe side effects such as cardiac hypertrophy, liver dysfunction, and masculinization in women have been described.231 Because of their (presumed) positive effects on muscle mass and function, the administration of AAS to athletes is prohibited at all times and both GC-MS(/MS) and LC-MS(/MS) approaches are routinely employed for their detection in doping control urine samples. But while synthetic AAS can easily be detected by using the abovementioned techniques, significantly more complex methodologies including the longitudinal monitoring of individual urinary steroid concentrations/ratios and IRMS need to be employed to provide evidence for the exogenous administration of pseudo-endogenous compounds.
Alternatively, the misuse of endogenous AAS can be proven by the detection of their intact esters in the athlete’s blood.232 Steroid esterification is a strategy to improve both the bioavailability and pharmacokinetic properties of the drug and as such conjugates can’t be synthesized by the human body, their presence in a doping control sample is a clear evidence for the exogenous origin. Following intramuscular administration, steroid esters are slowly released from the muscle tissue into the circulation, and the duration of this process depends on the length of the attached ester chain. In the blood, they are enzymatically cleaved by esterases, resulting in the release of the active compound.
Due to the rapid hydrolysis in the circulation, only plasma and serum can be used as biological matrices for the detection of intact steroid esters.233 However, the expected low plasma/serum concentrations necessitate sophisticated extraction and detection strategies to ensure utmost analytical sensitivity. Over the years, different assays have been developed, which combine protein precipitation and/or liquid-liquid-extraction (LLE) with GC- or LC-MS(/MS) following analyte derivatization.234 Overall, only three AAFs with testosterone esters have been reported so far.235 It can be assumed that the use of dried blood spots (DBS) (see 4.2) instead of plasma/serum could significantly improve the detectability of steroid esters, as the inactivation of esterases in the course of the drying process results in a significantly improved stability.236
3.2.6 Xenon
The noble gas xenon is a modern narcotic agent characterized by minimal side effects and favorable pharmacokinetic properties.237 Moreover, an organ-protective activity was described and it was found to stimulate EPO production through activation of the EPO transcription factor hypoxia-inducible factor 1 α (HIF-1α). For that reason, xenon has been reportedly used by elite athletes for performance-enhancement both before and during the 2014 Olympic Winter Games in Sochi, Russia.238 Even though it is still widely discussed if xenon inhalation actually results in a sustained improvement of athletic performance,239 it was added to section S2.2. (“Peptide hormones, growth factors, related substances and mimetics – HIF activators”) of the WADA Prohibited List in September 2014.240 As the noble gas has a high solubility in plasma and whole blood, these matrices were employed for the development of the first doping control detection assay based on GC time-of-flight (TOF) HRMS with headspace injection.241 Obviously, for a successful detection of xenon from plasma/blood, the sample containers have to be stored tightly closed at low temperatures (cooled or frozen), which further increases its solubility in the liquid phase. Moreover, sample transport and analysis should be conducted as quickly as possible and conditions applied to ABP blood specimens were found to be appropriate.242 Several studies indicate that the post-inhalation detection window in blood lies between 24 and 48 h.243 Alternatively, xenon can also be detected from urine, and also these specimens should be kept refrigerated or frozen.244
3.2.7 Further aspects
Targeted testing of urine and blood for the presence of doping agents requires information on their structure, mass spectrometric behavior, pharmacokinetic properties, elimination, and metabolic fate.245 Consequently, frequent switching of cheating athletes to new, unknown compounds represents an enormous challenge for anti-doping laboratories. Therefore, a novel, untargeted analytical strategy employing plasma as biological matrix, protein depletion for sample preparation, and LC-HRMS/MS for data acquisition was suggested.246 As plasma contains the circulating, unmodified parent drug, the use of this biological matrix allows for a comprehensive screening for both known and unknown compounds. And by using a high-resolution mass spectrometer in polarity switching mode and performing higher energy collision dissociation (HCD) fragmentation experiments without precursor ion pre-selection, comprehensive data for retrospective evaluation and the identification of emerging xenobiotics in the low ng/mL range can be generated. Another challenge represents the detection of substances prohibited only in-competition, such as stimulants, glucocorticoids and narcotics.247 Here, athletes are only sanctioned if pharmacologically relevant levels of a prohibited drug are detected in an in-competition doping control sample. Per definition,248 the time window of interest ranges from 11:59 pm on the day before the contest until its end or the collection of the doping control specimen. For selected doping agents prohibited in-competition only, urinary decision limits or reporting levels have been defined, however, several studies could demonstrate that urine samples have only a limited usability for the determination of effective in-competition blood concentrations of performance-enhancing drugs: Either, maximum blood concentrations are missed 249 or drug levels exceeding urinary thresholds correspond to (presumably) pharmacologically irrelevant blood concentrations.250 Thus, the analysis of blood instead of urine would provide more accurate data, but due to the abovementioned disadvantages (Sections 1 and 2), comprehensive in-competition blood sampling is rather difficult to realize. But complementary blood-derived matrices such as DBS and dried plasma spots (DPS) represent a very promising solution as their collection is less invasive and they are easier to handle (see also Section 4.2).
4 Future
4.1 Steroid profile
As indicated in Section 3.2.5, individual urinary steroid concentrations and concentration ratios are routinely monitored within the steroidal module of the ABP to identify athletes which might have misused pseudo-endogenous AAS such as testosterone for performance-enhancement.251 For confirmation purposes, the carbon isotopic ratios (13C/12C) of the detected steroids are determined by using IRMS, as they differ substantially between endogenous and synthetic steroids. However, urinary steroid markers can fail to detect the misuse of testosterone or its pro-hormones in athletes with a deletion/deletion (del/del) UGT2B17 genotype and women.252 The UGT2B17 gene encodes for the enzyme UDP-glucuronosyltransferase 2B17, which plays a central role in steroid metabolism.253 Individuals with a del/del genotype are characterized by a reduced testosterone glucuronidation and therefore low natural ratio of testosterone over epitestosterone (T/E), which is one of the key markers to uncover steroid misuse in sports. In women, urinary levels of testosterone and related steroid hormones are significantly lower than in men and subject to a large variation due to the menstrual cycle.254 Additionally, the adrenal, ovary, and extra-glandular testosterone produced in women doesn’t respond to an exogenous steroid administration with the same strong negative feedback as in men, where testosterone is predominantly synthesized in the testes. Consequently, changes in the female T/E ratio result only from high urinary testosterone levels caused by an exogenous administration of the steroid hormone and are not enhanced by a simultaneously reduced endogenous production of testosterone and its bio‐inactive epimer epitestosterone as in men. For that reason, the evaluation of biomarkers such as the T/E within the ABP can be significantly less effective in female athletes. Moreover, several other confounding factors such as bacterial contamination and ethanol consumption can influence urinary steroid concentrations.255
In such cases, the quantification of steroid hormones in plasma or serum specimens represents a valuable complementary analytical strategy and several studies already demonstrated that both a quantitative determination of endogenous steroid hormones from plasma/serum by using ultra-high-performance LC (UHPLC) and HRMS/MS,256 and a subsequent confirmation by multi-dimensional gas chromatography (MDGC) and IRMS 257 are possible. In the last years, several cases have been described where steroid doping could only be identified in female athletes by the detection of strong, transient elevations in serum testosterone concentrations and at the same time normal urinary T/E ratios.258 For that reason, both WADA and anti-doping laboratories are currently working on the short-term implementation of blood steroid profiling into the ABP.
4.2 Microsampling
For many years, the analysis of whole blood, plasma, and serum has been an indispensable part of routine sports drug testing. Nevertheless, the collection, transportation, and storage of these specimens require considerable efforts and costs (see also Sections 2 and 3). Whole blood and serum microsampling can overcome these limitations: Both DBS and DPS are derived from capillary blood obtained by a finger prick and spotted on a cellulose-based DBS sampling card.259 The sample collection procedure is minimally invasive, simple, quick, safe, relatively painless, and can be conducted by a regular DCO or the supervised athlete him-/herself. Due to the high analyte stability, the small sample volume (10-30 µL/spot), and the reduced biohazard risk, DBS and DPS can be shipped at ambient temperature in a comparably small tamper-evident sample container which contains a desiccant and protects the samples from light. In the anti-doping laboratory, storage at -20°C is advised, and due to the compact size of the sample containers, only little storage space is required.
During the last several years, the applicability of DBS to sports drug testing has been demonstrated for many different doping agents, comprising low molecular mass analytes such as AAS, stimulants and narcotics as well as proteins and peptides as for example ESAs and hGH.260 Moreover, measurements for the hematological module of the ABP has been made possible by measuring the hematocrit using near-infrared (NIR) spectroscopy and employing novel, protein-based biomarkers for the quantification of different types of blood cells.261 Especially for labile target analytes such as steroid esters and the peptide Synacthen, DBS were found to be of enormous value as the dehydratization during the drying process results in a reduced enzymatic activity and thus enhanced analyte stability.262 While an off-line DBS sample preparation can be a time-consuming process, both on-line approaches based on flow-through/unilateral fluidic assemblies and complex robotic systems for an automated DBS extraction and purification have been developed to allow for high-throughput sample analysis, minimize analyte losses during sample transfer, and reduce the risk for sample contamination.263
In consideration of all these benefits, DBS and DPS can substantially increase the testing frequency of athletes and thus availability of doping control specimens.264 This would not only improve monitoring of hormone/biomarker levels for ABP approaches, but also support decision making processes, especially regarding compounds prohibited in-competition only and in suspected cases of inadvertent doping.265 Additionally, they would facilitate testing of athletes in remote areas, the organization of large sport events, and doping tests in sport disciplines where the sport federations have not signed the World Anti-Doping Code (WADC) and only specific subsets of doping agents are relevant.266 However, there are also some limitations when using such microsamples for sports drug testing:267 The analytical sensitivity is reduced by the small sample volume and high matrix complexity, which results in short detection windows and a moderate retrospectivity. Moreover, only a limited number of biological replicates is available and the so-called hematocrit effect can complicate analyte quantification. Since the 2022 Olympic Winter Games in Beijing, the WADA has officially included DBS in their testing program for the detection of non-threshold substances and released a technical document to harmonize sample collection, transportation, storage, and analysis.268 But both laboratory performance levels and threshold values for the quantitative determination of certain doping agents yet have to be established.269
5. Summary
In many ways, blood plays an important role in sports drug testing: It can be misused by athletes to increase endurance performance and is the most important sample matrix to provide direct/indirect evidence for homologous/autologous blood doping. It is also an indispensable biological matrix for the analysis of hGH and HBOCs and can significantly improve the detection of certain ESAs, small peptide hormones such as insulin, endogenous AAS, and the noble gas xenon. Moreover, the use of blood can be of added value for the (retrospective) identification of emerging xenobiotics where information on the analyte’s metabolism is not (yet) available, and facilitate results interpretation in cases of AAFs with compounds prohibited only in-competition. In the near future, blood analysis will also complement steroid profiling and the use of blood microsamples such as DBS in addition to whole blood, plasma, and serum will significantly increase the testing frequency of athletes and thus improve various aspects of doping control analysis.
6. Acknowledgments
The authors thank the Manfred-Donike Institute for Doping Analysis (Cologne, Germany) and the Federal Ministry of the Interior, Community and Building (Berlin, Germany) for supporting the presented work.
About the Authors:
Dr. Katja Walpurgis is a senior scientist at the Center for Preventive Doping Research/Institute of Biochemistry of the German Sport University Cologne and her work focuses on the development of novel detection methods for performance-enhancing protein drugs by using different proteomics techniques such as gel electrophoresis, Western blotting, and LC-HRMS.
Dr. Andreas Thomas is a senior scientist at the Center for Preventive Doping Research/Institute of Biochemistry of the German Sport University Cologne. He is working in various fields of modern sports drug testing with focus on LC-HRMS and related techniques.
Prof. Mario Thevis graduated in organic chemistry and sports sciences before completing his PhD in Biochemistry in 2001. After post-doctoral research and senior scientist roles, he was appointed as Professor for Preventive Doping Research at the German Sport University Cologne in 2006. Mario Thevis further qualified as Forensic Chemist, and in 2017 he accepted the position of Director of the Institute of Biochemistry and WADA-accredited anti-doping laboratory of the German Sport University Cologne, Germany.