Introduction
Normocytic, normochromic anemia is among the most common laboratory abnormalities encountered in clinical practice, especially in older adults. In most cases, it reflects a hypoproliferative process in which the bone marrow is not producing red cells at a rate appropriate to the degree of anemia, as shown by an inappropriately low reticulocyte count. This tutorial focuses on that branch of the anemia tree: isolated, hypoproliferative, normocytic, normochromic anemia, the type often discovered incidentally and frequently mild or stable over time. The clinical task is to determine whether the finding points to a reversible cause, an organ-based process, or an underlying bone marrow disorder.
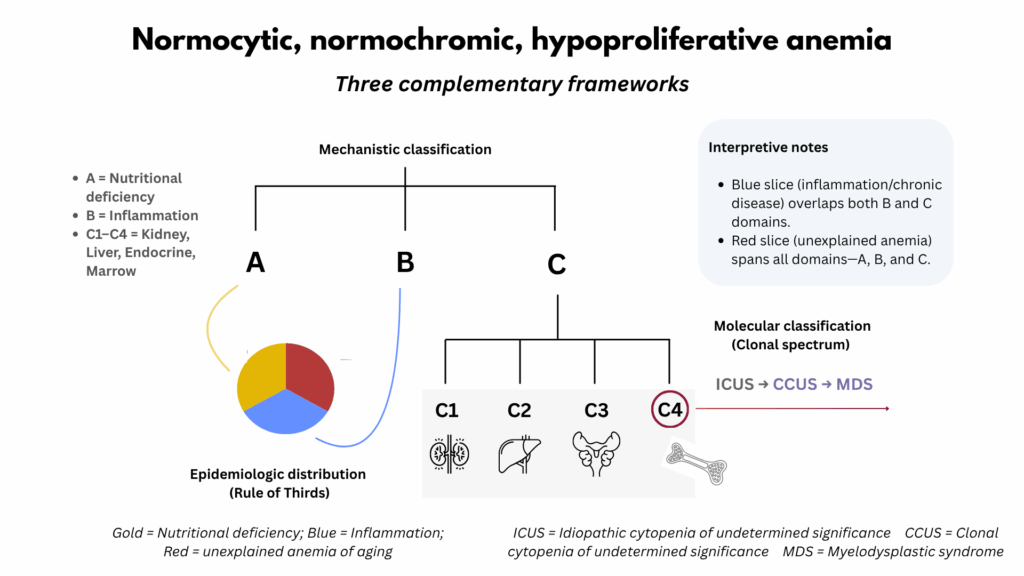
To approach this question systematically, three complementary frameworks can be applied. The A–B–C spine provides the clinical foundation, classifying hypoproliferative anemias according to their dominant mechanism: A for nutritional deficiency, B for inflammation, and C for organ dysfunction. The latter includes disorders of the kidney, liver, endocrine system, and bone marrow, organs that collectively support erythropoiesis. Conceptually, the A–B–C spine serves as both a functional and mechanistic framework, organizing the differential diagnosis by where along the erythropoietic pathway the process fails: at the level of substrate supply, inflammatory regulation, or organ-level support. In doing so, it links bedside reasoning with underlying biology and bridges the gap between epidemiologic and molecular models of anemia.
The anemia-of-aging model, derived from population studies, mirrors the A–B–C structure on a population level. About one-third of cases are due to nutritional deficiency, corresponding to A; one-third are associated with inflammation or chronic disease, corresponding to B and part of C; and one-third are labeled unexplained. This last group did not undergo routine bone marrow aspiration or biopsy in most studies and therefore likely includes primary marrow disorders as well as subclinical disturbances across nutrient supply, inflammatory regulation, and organ-level support of erythropoiesis that together encompass the A, B, and C of the spectrum.
The clonal spectrum (ICUS, CCUS, MDS) provides a molecular lens on the bone marrow compartment, which is one of the organ domains within C of the A–B–C framework. It identifies clonal hematopoiesis in a subset of otherwise unexplained cases localized to marrow pathology. This genetic perspective does not replace the clinical or epidemiologic models but refines the understanding of marrow-driven causes by revealing mechanisms that are not evident on peripheral blood or bone marrow morphology, or by standard laboratory testing.
Together, these three perspectives (clinical, epidemiologic, and biologic) form a layered model of hypoproliferative normocytic anemia. When considered together, they align bedside reasoning with population data and molecular insight, offering a more complete understanding of this common but complex condition.
In this tutorial, we will:
- Conclude by integrating all three approaches into a unified, layered model.
- Review the A–B–C spine, the practical diagnostic framework used in everyday clinical reasoning.
- Examine the anemia-of-aging model, which places hypoproliferative anemia in an epidemiologic context.
- Explore the clonality spectrum, which reframes otherwise unexplained cases in molecular terms.
- Layer these three schemes on top of one another to build a unified framework that links clinical reasoning, epidemiology, and biology.
Learning objectives
- Describe the major categories of hypoproliferative normocytic anemia using the A-B-C spine.
- Understand how the anemia-of-aging (UAA) framework relates to these categories.
- Recognize how modern genomics reframes “unexplained” anemia as a continuum from ICUS → CCUS → MDS.
- Integrate all three frameworks into a single cohesive model.
💡 After reading, you can check your understanding with a short self-assessment quiz located at the end of this tutorial.
Podcast
An AI-generated conversation exploring the clinical approach to normocytic, normochromic anemia through the A–B–C spine, the Rule of Thirds, and the Clonal Spectrum frameworks.
Case Vignette
Mr. J is a 74-year-old man referred for evaluation of mild, stable normocytic anemia (Hb 10.8 g/dL). He feels well, with no bleeding, constitutional symptoms, or medication changes. Kidney and liver function are normal. Iron, B12, and folate studies are within range. The reticulocyte count is low. White cell and platelet counts are normal.
| Test | Result | Reference Range |
|---|---|---|
| Hemoglobin | 10.8 g/dL | 13–17 |
| MCV | 91 fL | 80–100 |
| Reticulocyte count | 0.6 % | 0.8–2.5 |
| WBC | 5.2 × 10⁹/L | 4–11 |
| Platelets | 235 × 10⁹/L | 150-400 |
| Ferritin | 85 µg/L | 30–300 |
| Vitamin B12 | 420 pg/mL | 200–900 |
| Creatinine | 0.9 mg/dL | – |
| CRP | < 5 mg/L | – |
| Additional tests | Normal liver, thyroid, and testosterone levels; normal SPEP | – |
Values representative of the initial evaluation.
Clinical Preface: When the Work-Up Is Normal
At first glance, this is the kind of anemia most clinicians approach using a familiar stepwise framework, starting with nutritional causes, then considering inflammation, and finally evaluating organ function. We’ll formalize this approach later as the A–B–C spine, the first of three complementary frameworks explored in this tutorial.
In many patients like Mr. J, every reasonable peripheral blood test aligned with that basic framework has already been done — and all results are normal. Nutritional deficiencies are excluded, inflammatory markers are negative, and kidney, liver, and endocrine function are intact. What remains is an anemia that defies straightforward classification. Among adults over sixty-five, this scenario is common: a mild, stable, normocytic anemia with no apparent cause. At that juncture, the only unperformed test is often a bone marrow aspiration and biopsy. In practice, we frequently defer that step, knowing that the yield for uncovering a serious hematologic disorder is low and that the result would rarely change management.
We are thus left acknowledging that, despite a thorough evaluation, the cause of Mr. J’s anemia remains uncertain. In the sections that follow, we’ll broaden our view through frameworks that may not provide an immediate diagnosis but will deepen our understanding as clinicians. They remind us that diagnostic uncertainty often reflects the limits of our current tools rather than a failure of reasoning, and that beneath these quiet anemias lie the subtle biologic changes of aging hematopoiesis.
→ To understand where Mr. J fits within these perspectives, we next turn to the formal definition of the three frameworks that structure this tutorial.
The A–B–C Spine
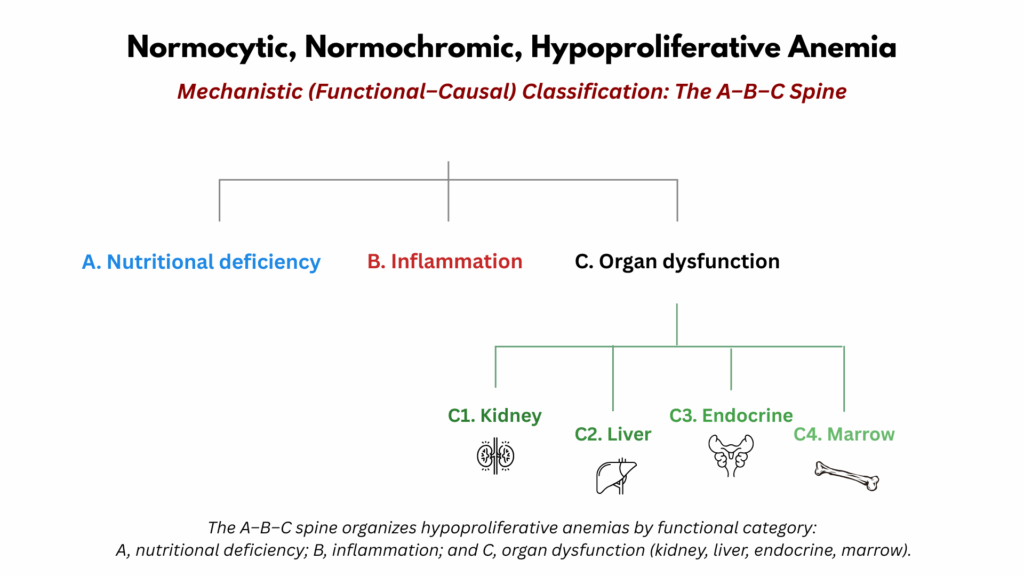
The A–B–C spine provides a mechanistic framework for understanding hypoproliferative, normocytic, normochromic anemia. Rather than classifying by disease, it organizes the differential diagnosis around three physiological domains required for normal red cell production: A) substrate supply (nutrients such as iron, folate, and vitamin B12), B) iron mobilization and utilization (often disrupted by inflammation and altered hepcidin signaling), and C) regulatory control and marrow responsiveness (dependent on hormonal, renal, hepatic, endocrine, and marrow function).3 Each domain represents a distinct point along the erythropoietic pathway where production may falter. The purpose of the A–B–C spine is not to replace disease-based classification but to encourage clinicians to think physiologically—identifying which component of the erythropoietic system is failing and why.
- Nutritional Deficiency:
- Definition and Features:
- Although nutritional anemias are classically microcytic (iron deficiency) or macrocytic (vitamin B₁₂ or folate deficiency), a substantial proportion of patients with true deficiency may appear normocytic, either early in the course or because of coexisting factors that shift the mean corpuscular volume (MCV) toward normal.
- Normocytic presentation may occur when:
- Deficiency is early or partial, before full morphologic expression develops.
- Mixed populations of small and large red cells yield a “normal” average MCV.
- Opposing influences on cell size (e.g., iron deficiency + macrocytosis from alcohol or liver disease) balance each other out.
- Mechanisms and Representative Deficiencies:
- Iron deficiency:
- Pattern: Typically microcytic but may be normocytic.
- Explanation: AThe MCV may be normal early in the course, when microcytosis is only beginning to develop, or when microcytic cells coexist with larger populations of normocytic or macrocytic cells. The latter scenario can occur with concurrent vitamin B₁₂ or folate deficiency, liver disease, alcohol use, or pregnancy.4.
- Testing: Evaluate with serum ferritin and transferrin saturation, recognizing that ferritin may be spuriously elevated in inflammatory states.
- Treatment: Oral or intravenous iron depending on tolerance, absorption, and urgency of repletion; address the underlying cause of loss or malabsorption.
- Vitamin B₁₂ or folate deficiency:
- Pattern: Typically macrocytic, but may be normocytic.
- Explanation: A normocytic presentation may occur early in the course, before macrocytosis develops, or when concurrent microcytic processes—such as iron deficiency, thalassemia trait, or anemia of inflammation—counterbalance the increase in cell size.
- Testing: Measure serum vitamin B₁₂ and folate levels; if results are borderline, assess methylmalonic acid and homocysteine for confirmation.
- Treatment: Replace the deficient vitamin (parenteral or high-dose oral B₁₂; oral folate) and address underlying causes such as malabsorption, dietary insufficiency, or medication effects. Correct coexisting deficiencies as needed.
- Copper deficiency:
- Pattern: Typically normocytic or mildly macrocytic, often accompanied by neutropenia.
- Explanation: Copper deficiency reduces effective red cell production and may mimic iron deficiency through impaired iron utilization, even though microcytosis is absent.
- Testing: Low serum copper and ceruloplasmin; consider in patients with malabsorption (especially post–bariatric surgery), prolonged parenteral nutrition, or unexplained anemia unresponsive to iron therapy.
- Treatment: Copper repletion (oral or intravenous, depending on severity). Identify and correct contributing factors such as excessive zinc intake. Hematologic recovery usually occurs within weeks.
- Iron deficiency:
- Population Perspective:
- Nutritional deficiencies remain major global contributors to anemia, though in developed settings their relative share is smaller.
- In general adult populations:
- Iron deficiency accounts for a large proportion of mild normocytic or microcytic anemias, especially in menstruating women and those with occult GI loss.
- Vitamin B₁₂ and folate deficiency become more prevalent with aging, malnutrition, or malabsorption.
- Copper deficiency is uncommon but increasingly recognized in postoperative and supplement-related settings.
- Individual Patient Perspective:
- At the bedside, the challenge is to recognize nutritional deficiency even when MCV is normal.
- Check iron indices (ferritin, transferrin saturation), noting that inflammation may obscure classic patterns.
- Assess B₁₂ and folate in normocytic anemia with a wide RDW or with neurologic or mucocutaneous clues suggesting deficiency
- Consider copper deficiency in patients with unexplained anemia and neutropenia after gastric surgery or on zinc supplements.
- As in all hypoproliferative states, the reticulocyte count is low but should rise briskly within 7–10 days of correction.
- At the bedside, the challenge is to recognize nutritional deficiency even when MCV is normal.
- Definition and Features:
- Inflammation:
- Definition and Features:
- Inflammatory disorders reduce red cell production through cytokine-mediated pathways that impair iron mobilization and blunt erythropoietin (EPO) responsiveness.
- The resulting anemia of inflammation (AI)—formerly anemia of chronic disease—is typically normocytic, normochromic, and hypoproliferative, with an inappropriately low reticulocyte count for the degree of anemia.
- AI occurs across a spectrum: from classic (overt) inflammatory states such as infection, autoimmune disease, and cancer, to low-grade, subclinical inflammation seen with aging, obesity, or metabolic syndrome (“inflammaging”).
- Mechanisms:
- Classic (overt) anemia of inflammation:
- Definition: Established form of inflammation-associated anemia occurring in chronic infection, autoimmune disease, malignancy, or metabolic disorders.
- Pathophysiology:
- ↑ Hepcidin → ↓ iron release from macrophages and enterocytes.
- ↓ EPO production and ↓ marrow responsiveness.
- Shortened RBC lifespan due to macrophage activation and oxidative stress.
- Laboratory pattern:
- Low serum iron and transferrin saturation.
- Normal or elevated ferritin.
- Low reticulocyte count.
- Normal or mildly decreased EPO levels.
- Context:
- Occurs in a wide spectrum of chronic inflammatory conditions — such as rheumatoid arthritis, inflammatory bowel disease, chronic infection, and cancer — but can also appear transiently during prolonged acute illness.
- The anemia evolves gradually as inflammatory cytokines blunt erythropoietin responsiveness and restrict iron availability.
- Significance
- The prototype of inflammatory anemia, serving as the mechanistic reference for subclinical variants.
- Important to recognize because it reflects disease activity rather than iron deficiency and typically resolves when inflammation abates.
- Low-grade (subclinical) inflammation:
- Definition: Mild, chronic inflammatory activity that does not meet criteria for overt inflammatory disease but exerts measurable effects on erythropoiesis and iron metabolism.
- Contexts:
- Older adults: Often labeled anemia of aging, reflecting modest elevations in inflammatory cytokines (IL-6, TNF-α) associated with immunosenescence, comorbidity burden, and physiologic decline in marrow responsiveness and renal EPO output.
- Metabolic syndrome and obesity: Characterized by adipose-derived cytokine production and hepatic hepcidin upregulation, leading to impaired iron mobilization and relative EPO resistance in otherwise younger or middle-aged adults.
- Mechanism:
- Same cytokine–hepcidin axis as classic AI but lower intensity, often with normal CRP or ESR.
- ↑ Hepcidin and iron sequestration (common to both settings).
- ↓ Erythropoietin production or responsiveness (more prominent in the elderly).
- Direct cytokine-mediated suppression of erythroid progenitors (variable contribution).
- Key Point: Although both groups show similar laboratory profiles—normocytic or mildly microcytic anemia with low serum iron and low transferrin saturation—the underlying biology and clinical context differ. Distinguishing them helps avoid overgeneralization of “low-grade inflammation” as a single category.
- Classic (overt) anemia of inflammation:
- Population Perspective:
- Across populations, anemia of inflammation (AI) is one of the most common causes of normocytic, normochromic, hypoproliferative anemia.
- It occurs in both acute and chronic inflammatory states—infectious, autoimmune, malignant, or metabolic.
- Epidemiologic studies show a strong correlation between inflammatory burden and prevalence of anemia, even in subclinical conditions such as obesity, metabolic syndrome, and aging (“inflammaging”).
- Typical laboratory trends across cohorts include:
- Low serum iron and transferrin saturation
- Normal or elevated ferritin
- Low reticulocyte count
- Normal or mildly decreased EPO levels
- Together, these features reflect the hepcidin-mediated restriction of iron availability and cytokine suppression of erythropoiesis that define AI at the population level.
- Individual patient perspective:5
- In any given patient, however, the expression of anemia is highly variable:
- Some patients with marked systemic inflammation maintain normal hemoglobin, while others develop profound anemia with only modest cytokine elevation.
- The degree of anemia does not correlate linearly with CRP, ESR, or hepcidin.
- Coexisting factors—iron deficiency, renal insufficiency, endocrine disorders, chronic infection, or neoplastic disease—modify both severity and morphology.
- In milder “low-grade” or “subclinical” inflammation, the anemia may be stable, normocytic, and only 1–2 g/dL below normal, making it indistinguishable from the so-called unexplained anemia of aging
- In any given patient, however, the expression of anemia is highly variable:
- Definition and Features:
Historical Note: From “Anemia of Chronic Disease” to “Anemia of Inflammation”
Historically, the term “anemia of chronic disease (ACD)” was used to describe the mild, normocytic, normochromic anemia accompanying chronic infections, autoimmune disease, cancer, and CKD.
- The word chronic implies duration rather than mechanism, and the same pathophysiology can occur acutely (e.g., sepsis, postoperative inflammation).
- “Inflammation” better captures the underlying biology (IL-6 → hepcidin → iron sequestration → impaired erythropoiesis).
Major reviews now use “anemia of inflammation” (AI) almost exclusively.
- Organ Dysfunction – Organ failure impairs erythropoiesis through multiple mechanisms: reduced hormonal support, altered metabolic milieu, or direct marrow suppression.
- C1. Kidney
- Definition and Features:
- Anemia of chronic kidney disease (CKD) is a normocytic, normochromic, hypoproliferative anemia that develops as renal function declines, affecting roughly one-third of patients with stage 3 disease and the majority of those with stage 5.
- It reflects impaired erythropoietin (EPO) production and a reduced marrow response to EPO in the setting of uremia, inflammation, and altered iron metabolism.
- Severity generally increases with CKD stage but varies widely among individuals, emphasizing the interplay of EPO deficiency, inflammation, and iron restriction rather than a single deterministic relationship between hemoglobin and creatinine.
- Mechanisms:
- ↓ Erythropoietin (EPO) production by hypoxia-sensitive/responsive peritubular fibroblast-like cells.
- EPO resistance due to chronic inflammation, uremic milieu, and oxidative stress.
- Iron-restricted erythropoiesis from elevated hepcidin and blood losses from dialysis or phlebotomy.
- Shortened RBC lifespan (mild contribution).
- Population Perspective:
- Across populations, there is a strong, graded association between kidney function and hemoglobin level.6
- Large cohort studies (e.g., NHANES, CKD registries) show that mean hemoglobin declines progressively as eGFR falls, particularly below ~60 mL/min/1.73 m².
- Individual Patient Perspective:7
- For any given individual, hemoglobin at a single time point cannot be mapped to a specific creatinine or eGFR.
- The relationship is broadly stochastic, shaped by EPO production, iron status, inflammation, comorbidities, and marrow responsiveness.
- Two patients with the same eGFR may have very different hemoglobin values.
- One patient’s hemoglobin may remain stable over a range of eGFR values before dropping once EPO reserve is exceeded.
- Thus, while the population trend is predictable, the timing and degree of anemia in a given patient are not—making longitudinal trends more informative than snapshots.
- Definition and Features:
- C2. Liver
- Definition and Features:
- Although macrocytosis is a well-known feature of anemia in liver disease—occurring in up to half of cases, particularly those related to alcohol use—normocytic anemia remains common, especially in viral, cholestatic, and nonalcoholic liver disease.
- The anemia is most often hypoproliferative, reflecting blood loss with secondary iron deficiency, nutritional deficiency, alcohol toxicity, and inflammation.
- However, hyperproliferative patterns may occur in specific settings—such as acute or chronic blood loss (before iron depletion develops), spur cell anemia, or hypersplenism—where increased red-cell turnover drives a compensatory marrow response.
- Mechanisms:
- Common indirect causes (These account for most anemia in chronic liver disease):8
- Gastrointestinal blood loss:
- From portal hypertensive gastropathy, varices, peptic ulcer disease, gastritis, hemorrhoids, or coagulopathy.
- Nutritional deficiencies:
- Folate deficiency (especially in alcohol-associated liver disease).
- Iron deficiency secondary to chronic GI blood loss or poor diet.
- Alcohol toxicity:
- Direct marrow suppression, folate deficiency, and macrocytosis (often even without anemia).
- Gastrointestinal blood loss:
- Liver disease–specific (intrinsic) factors (though less common, these reflect direct hepatic involvement in erythropoiesis or red-cell turnover):9
- Hypersplenism (splenomegaly from portal hypertension):
- Hypersplenism from portal hypertension may contribute to cytopenias by sequestering blood cells, but in isolation it rarely causes anemia, because the bone marrow typically compensates by increasing erythropoiesis. When anemia occurs, it usually reflects impaired marrow response (from alcohol toxicity, inflammation, or deficiency) or coexisting hemolysis.
- Spur cell (acanthocytic) anemia:
- In advanced cirrhosis, altered RBC membrane lipid composition → reduced deformability → hemolysis.
- Occurs in a small subset (<5%) of patients with end-stage liver disease.
- Anemia of inflammation (hepcidin-mediated iron restriction):
- Chronic hepatic inflammation may contribute to an anemia of inflammation phenotype (normocytic, low serum iron, high ferritin).
- Hepatitis:
- Hepatitis-associated aplastic anemia is considered a variant of aplastic anemia.
- Usually seronegative (etiological agent unknown).
- Hypersplenism (splenomegaly from portal hypertension):
- Common indirect causes (These account for most anemia in chronic liver disease):8
- Population Perspective:
- Cross large cohorts, anemia is common in chronic liver disease, affecting 30–70% of patients depending on etiology and severity.
- Macrocytosis occurs in roughly half of those with alcohol-related disease but is less frequent in viral or cholestatic causes.
- The prevalence and severity of anemia increase with worsening hepatic function (Child–Pugh or MELD stage).
- The distribution of anemia types differs by cause—blood loss and iron deficiency predominate in bleeding-related cases, while macrocytosis dominates in alcohol-related disease.
- Individual Patient Perspective:
- The morphology (macrocytic vs. normocytic) and proliferative pattern (hypo- vs. hyper-) vary by etiology and comorbid factors—e.g., nutritional status, alcohol exposure, splenomegaly, and inflammation.
- Hypersplenism and blood loss can both produce hyperproliferative anemia, provided marrow reserve and iron availability are intact.
- Spur cell anemia is a rare,10 hyperproliferative hemolytic process signaling advanced hepatic dysfunction and poor prognosis.
- In many patients, anemia is multifactorial—part bleeding, part deficiency, part inflammation.
- Trends over time in hemoglobin, MCV, and reticulocyte count often provide the most insight into the dominant mechanism.
- Definition and Features:
- C3. Endocrine
- Definition and Features
- Endocrine disorders represent an uncommon but clinically important cause of normocytic, normochromic, hypoproliferative anemia.
- They include hypothyroidism, hypogonadism, adrenal insufficiency, and pituitary insufficiency—each capable of blunting erythropoiesis through distinct hormonal mechanisms.
- The anemia is usually mild, stable, and reversible with hormone replacement, underscoring the endocrine system’s role as a permissive regulator of red cell production.
- Collectively, endocrine causes account for <5% of normocytic anemias once nutritional, inflammatory, and renal etiologies are excluded.
- Mechanisms
- Hypothyroidism12
- Pattern: Most often normocytic and hypoproliferative, though macrocytosis is common; anemia occurs in ~30–40% of overt cases.
- Mechanisms:
- ↓ Oxygen consumption → ↓ renal EPO synthesis.
- ↓ Bone marrow proliferation and RBC turnover.
- Nutritional deficiency (B₁₂, folate, or iron) may coexist, accentuating anemia or altering MCV.
- Notes: Corrects with thyroxine replacement; persistent anemia warrants reassessment for another cause
- Hypogonadism
- Pattern: Normocytic, hypoproliferative anemia; common in older men and those with chronic illness.
- Mechanisms:
- Testosterone stimulates erythropoiesis both directly (marrow androgen receptors) and indirectly (increased EPO).
- Low testosterone → reduced EPO drive → mild anemia.
- Notes: Testosterone replacement raises hemoglobin by ~1–2 g/dL on average; excessive therapy can cause erythrocytosis.
- Adrenal Insufficiency
- Pattern: Usually normocytic and mild; sometimes accompanies other cytopenias.
- Mechanisms:
- Glucocorticoids enhance marrow responsiveness to EPO; deficiency blunts this effect.
- ↓ Cortisol may also decrease EPO production directly.
- Notes: Anemia improves with glucocorticoid replacement; persistent cytopenias may suggest panhypopituitarism or autoimmune overlap.
- Pituitary Insufficiency
- Pattern: Normocytic anemia, sometimes with pancytopenia in panhypopituitarism.
- Mechanisms:13
- Deficiency of ACTH (→ low cortisol) and GH reduces EPO output and marrow drive.
- Secondary hypogonadism contributes further to erythropoietic suppression.
- Notes: Corrects with targeted hormone replacement (hydrocortisone, levothyroxine, sex steroids, GH).
- Hypothyroidism12
- Population Perspective
- Endocrine causes account for a small fraction (<5%) of hypoproliferative anemias in epidemiologic studies.
- Hypothyroidism:
- The most frequent contributor, especially among women and older adults.
- Reported prevalence of anemia in patients with hypothyroidism 20%-60%.14
- Hyperthyroidism:
- Reported prevalence of anemia in patients with hyperthyroidism 10%-25%.15
- Hypogonadism-related anemia is underrecognized in aging men but increasingly noted in population cohorts.
- Prevalence of anemia 29.6% in prospective cohort study of 58 men with hypogonadism (serum testosterone level < 2.35 ng/ml).16
- Adrenal and pituitary insufficiency are rare, typically seen in tertiary or complex endocrine disease.
- prevalence of anemia 35% in men with newly diagnosed macroprolactinoma.18
- Across studied cohorts of unexplained cytopenias, marrow failure and early clonal disorders represent a small but clinically important subset.
- In population studies of “unexplained anemia of aging,” up to one-third of idiopathic cases harbor clonal hematopoiesis when next-generation sequencing is performed.
- The prevalence of CHIP (clonal hematopoiesis of indeterminate potential) increases with age (>10 % after age 70), and a subset progress to CCUS or MDS over years.
- Thus, the boundary between “anemia of aging” and “incipient marrow disease” is porous and dynamic.
- Individual Patient Perspective:
- In a given patient, distinguishing primary marrow failure from secondary hypoproliferation (e.g., kidney, endocrine, inflammation) requires integration of:
- Reticulocyte index (inappropriately low).
- Other cytopenias (suggestive of broader marrow involvement).
- Peripheral smear (dysplasia, blasts, teardrops).
- Serum studies (iron, B12, folate, EPO).
- And, when indicated, bone marrow biopsy and molecular testing.
- In a given patient, distinguishing primary marrow failure from secondary hypoproliferation (e.g., kidney, endocrine, inflammation) requires integration of:
- In many older adults with mild, stable, normocytic anemia and otherwise normal labs, marrow biopsy is deferred because it seldom changes management.
- In patients with progressive cytopenias, macrocytosis, or abnormal smears, marrow evaluation may reveal ICUS, CCUS, or overt MDS.
- The decision to pursue invasive testing must weigh diagnostic yield vs. clinical impact, recognizing that many early clonal states remain indolent.
- prevalence of anemia 35% in men with newly diagnosed macroprolactinoma.18
Anemia of Aging
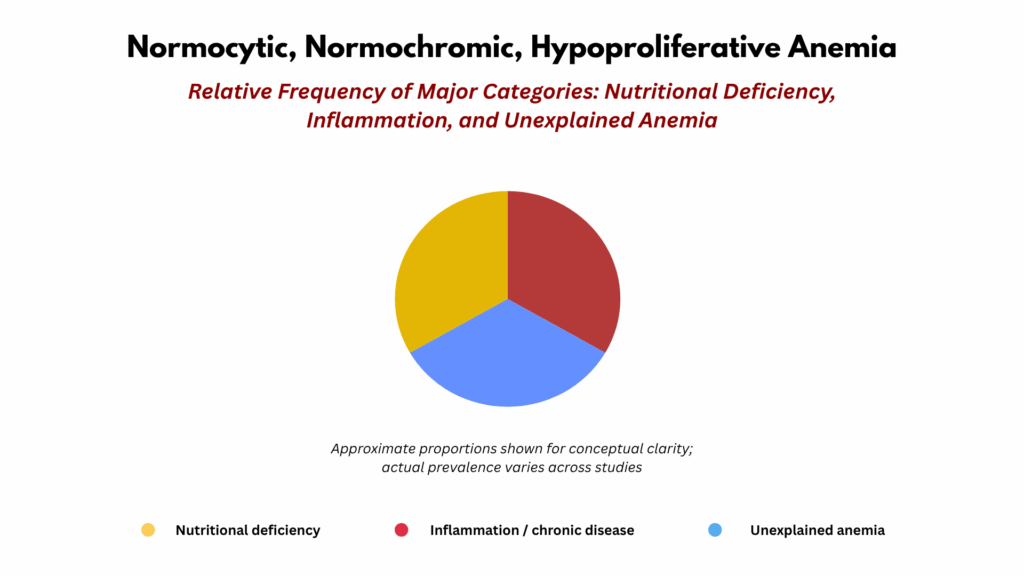
Population studies of anemia in older adults have shown a remarkably consistent pattern: about one-third of cases are due to nutritional deficiency, one-third to inflammation or chronic disease, and one-third remain unexplained.19 This “rule of thirds” applies to cohorts evaluated through peripheral blood testing but not through routine bone marrow aspiration or biopsy. As a result, the “unexplained” category almost certainly includes undiagnosed bone marrow–based processes, along with subtle nutritional, inflammatory, or organ-related abnormalities that fall below the resolution of current diagnostic tools. This simple pie chart therefore highlights both what we know and what we still miss in the evaluation of anemia in the elderly.

Figure 2. The Rule of Thirds in Anemia of Aging. The Rule of Thirds framework summarizes the distribution of anemia causes among older adults (typically ≥ 65 years). Approximately one-third of cases reflect nutritional deficiency (iron, folate, or vitamin B₁₂), one-third arise from chronic disease or inflammation, and one-third remain unexplained despite standard evaluation. This epidemiologic breakdown provides an entry point for mechanistic interpretation through the ABC spine: nutritional anemia aligns with Axis A, inflammatory anemia with Axis B, and unexplained anemia may involve subtle organ or marrow factors along Axis C. In hematologic studies, aging usually refers to adults 65 years and older, though age-related changes in erythropoiesis may begin earlier. The term does not imply frailty or disease, but rather the gradual biologic transition that accompanies advancing age—reduced marrow reserve, subtle erythropoietin resistance, low-grade inflammation (“inflammaging”), and increasing clonal hematopoiesis. When we speak of anemia of aging, we mean anemia in this population that cannot be attributed to identifiable nutritional, inflammatory, renal, endocrine, or malignant causes.
The Rule of Thirds Framework (Classification of Anemia in the Elderly)
- Nutritional deficiency (~⅓):
- Primarily iron, B12, and folate deficiency.
- Inflammation and chronic disease (~⅓):
- Includes overt inflammatory states (infection, autoimmune disease, malignancy), systemic disorders with inflammatory components such as chronic kidney or liver disease, and the low-grade inflammation of aging (“inflammaging”).20
- Unexplained anemia of aging (~⅓):
- A residual category defined by exclusion of known causes.
- Encompasses subtle renal or endocrine impairment, blunted erythropoietin response, and early or indolent marrow/clonal changes.
The first two categories—nutritional deficiency and inflammation—correspond closely to A and B of the A–B–C spine. The third, unexplained anemia of aging, likely represents a heterogeneous mix of subtle or overlapping processes that span the A–C continuum. These may include mild nutritional insufficiency, low-grade inflammation, diminished erythropoietin production or responsiveness, early or indolent marrow changes, and other forms of minor organ dysfunction that escape detection by standard testing. In this sense, the “unexplained” segment of the pie chart highlights not a single hidden cause but the limits of current diagnostic resolution. It reminds us that a substantial proportion of older adults with anemia remain unclassified when evaluated only by peripheral blood studies, revealing a diagnostic blind spot that the molecular perspective of the clonal spectrum begins to address.
The Unexplained Third: Unexplained Anemia of Aging
- Definition and Features:
- Unexplained anemia of aging (UAA) refers to anemia in older adults that persists after exclusion of nutritional deficiency, renal disease, chronic inflammation, endocrine dysfunction, and overt marrow disorders.
- It is typically mild (Hb 10–12 g/dL), normocytic, and hypoproliferative.
- UAA represents a diagnosis of exclusion and a window into the biologic effects of aging on erythropoiesis.
- Mechanisms (Proposed):
- The pathogenesis of unexplained anemia of older adults (UAOA) is multifactorial, reflecting both intrinsic bone marrow aging and extrinsic systemic factors:
- Reduced erythropoietic drive:
- Lower-than-expected erythropoietin (EPO) production for the degree of anemia
- Progressive resistance of erythroid progenitors to EPO stimulation due to stem cell aging and altered marrow signaling
- Subclinical (“inflammaging”) milieu:
- Chronic low-grade inflammatory cytokine activity (IL-6, TNF-α) with variable hepcidin upregulation
- Conflicting data on hepcidin levels in UAOA cohorts21
- Early clonal hematopoiesis:
- Presence of somatic mutations in hematopoietic cells (CCUS or CHIP) without morphologic dysplasia
- ~10–15% of UAOA cases ultimately reclassified as early myelodysplastic syndrome22
- Subtle nutritional or hormonal insufficiency:
- Marginal iron, B12, or vitamin D levels without overt deficiency
- Androgen deficiency in older men may increase hepcidin and blunt iron mobilization
- Marrow microenvironment aging:
- Senescence-associated changes in stromal and vascular niches impair erythropoiesis and red cell survival
- Reduced erythropoietic drive:
- The pathogenesis of unexplained anemia of older adults (UAOA) is multifactorial, reflecting both intrinsic bone marrow aging and extrinsic systemic factors:
- Population perspective:
- Accounts for roughly one-third of anemia in older adults after exclusion of nutritional, inflammatory, renal, and endocrine causes.
- Prevalence:
- Typically mild (Hb 10–12 g/dL), normocytic, and stable over time.
- Individual-Patient Perspective:
- Expression varies: some remain stable for years, others slowly decline.
- No single biomarker (EPO, CRP, ferritin) maps predictably to Hb level—just as eGFR doesn’t map cleanly to Hb in CKD.
- Distinguish stable, mild idiopathic anemia from evolving marrow disease (rising MCV, new cytopenias, dysplasia).
- Clinical Significance:
- Even mild UAA is linked to frailty, falls, hospitalization, and mortality.
- Causality uncertain—may reflect underlying biologic aging rather than direct driver.
- Monitor trends rather than treating numbers.
- Spectrum and Overlap:
- UAA likely shares biological territory with early marrow or clonal conditions such as ICUS and CCUS, which may precede myelodysplastic syndromes (MDS).
- These pre-MDS states are discussed in the next section.
The Clonal Spectrum (ICUS–CCUS–MDS Continuum)
The third and final framework considers anemia that arises from intrinsic marrow processes, conditions defined not by deficiency or inflammation but by acquired somatic mutations and disordered hematopoiesis. This spectrum extends from idiopathic cytopenia of undetermined significance (ICUS), through clonal cytopenia of undetermined significance (CCUS), to myelodysplastic syndromes (MDS).26 Each represents a stage along a continuum of clonality, dysplasia, and clinical consequence, bridging the gap between otherwise unexplained anemia and overt marrow failure.
This framework focuses squarely on the bone marrow, one of the four organ systems within C (organ dysfunction) of the A–B–C spine. It is therefore not a comprehensive model of anemia but rather a molecular lens directed at a single organ. Many cases within this spectrum, especially ICUS and CCUS, are not evident on routine bone marrow morphology or biopsy. They are revealed only through molecular profiling, which shows that the marrow can harbor clonal or pre-dysplastic changes beyond the reach of conventional pathology. In this way, the clonal spectrum refines our understanding of marrow-based anemia, complementing rather than replacing the broader clinical and epidemiologic frameworks.
Classification within the Clonal Spectrum
- ICUS (Idiopathic Cytopenia of Undetermined Significance):
- Persistent cytopenia(s) without clonal mutations or morphologic dysplasia.
- Represents “unexplained” cytopenia after full evaluation.
- CCUS (Clonal Cytopenia of Undetermined Significance):
- Cytopenia(s) with one or more somatic myeloid-type mutations but without definitive dysplasia or MDS-defining criteria.
- A clonal but sub-diagnostic state.
- MDS (Myelodysplastic Syndromes / early MDS):
- Cytopenia(s) with morphologic dysplasia and/or specific cytogenetic or molecular abnormalities meeting WHO criteria.
- Represents the clinically manifest phase of the spectrum.
Together, these entities represent a stepwise continuum from idiopathic cytopenia to clonal cytopenia to overt marrow failure.
Figure 3. The Clonal Cytopenia Spectrum: ICUS → CCUS → MDS. The clonal spectrum illustrates the continuum from idiopathic cytopenia of undetermined significance (ICUS) through clonal cytopenia of undetermined significance (CCUS) to myelodysplastic syndrome (MDS). ICUS represents persistent cytopenia without clonal mutations or dysplasia. CCUS introduces somatic mutations but lacks morphologic change. MDS combines clonality with dysplasia, often showing progression and clinical consequence. The left-to-right gradient reflects increasing clonal burden, genomic complexity, and risk of malignant evolution, providing context for marrow-based interpretations of unexplained anemia. Definition and Features
Viewed along a continuum:
- ICUS defines persistent cytopenia without identifiable cause, mutation, or dysplasia:
- No evidence of peripheral cytopenias.
- Criteria for diagnosis of MDS is not fulfilled.
- No MDS-related mutation as detected by conventional cytogenetics, FISH, or sequencing studies.
- ≥ 10% dysplasia of neutrophilic, erythroid, and/or megakaryocytes.
- < 5% blast cells detected by morphologic examination of the blood and/or bone marrow smears.
- CCUS adds the presence of one or more somatic mutations in myeloid-associated genes (e.g., DNMT3A, TET2, ASXL1) at a variant allele fraction ≥2%, but without morphologic evidence of MDS:
- Any degree of peripheral cytopenia lasting ≥ 4 months.
- Criteria for diagnosis of MDS is not fulfilled.
- Presence of ≥ 1 MDS-related mutation as detected by conventional cytogenetics, FISH, or sequencing studies.
- < 10% dysplasia of neutrophilic, erythroid, and/or megakaryocyte lineages.
- < 5% blast cells detected by morphologic examination of the blood and/or bone marrow smears.
- MDS is marked by clonal hematopoiesis plus morphologic dysplasia, cytogenetic abnormalities, or specific molecular signatures that confer diagnostic weight.
All three share the feature of ineffective hematopoiesis, reflected clinically by stable or progressive cytopenias—often anemia—and variably elevated MCV or RDW. Their distinction lies in clonality and morphologic consequence, not in immediate clinical presentation.
Proposed Mechanisms
- Genetic: Stepwise accumulation of somatic mutations in epigenetic, splicing, and DNA repair genes (DNMT3A, TET2, ASXL1, SF3B1, TP53).
- Cellular: Impaired self-renewal and differentiation of hematopoietic stem and progenitor cells.
- Microenvironmental: Aging marrow niche, altered stromal and cytokine signaling, and inflammatory stress (“inflammaging”) that selects for clonal advantage.
- Functional consequence: Reduced red cell output, dyserythropoiesis, and ineffective hematopoiesis.
Population Perspective
- ICUS and CCUS are predominantly identified in older adults undergoing marrow evaluation for persistent, otherwise unexplained cytopenias.
- The prevalence of clonal hematopoiesis increases sharply with age: by age 70, up to 10–20% of individuals harbor detectable mutations, though only a minority develop cytopenias.
- In population studies, CCUS carries a substantial risk of progression (approximately 10–30% over 5 years), intermediate between CHIP and overt MDS.
- MDS incidence rises from <5 per 100,000 at age 50 to >50 per 100,000 after age 80.
Individual Patient Perspective
- Patients may present with mild, stable cytopenia or slowly progressive anemia without overt dysplasia.
- Distinguishing ICUS from early CCUS requires next-generation sequencing, which reveals subclinical clonality not visible morphologically.
- Clinical trajectory is heterogeneous:
- ICUS: often indolent, may remain static for years.
- CCUS: higher risk of evolution to MDS or AML, depending on mutation type, number, and VAF.
- MDS: established marrow failure with potential for leukemic transformation.
- Management is largely observational unless cytopenias worsen or symptomatic anemia demands intervention.
Clinical Significance
- Recognition of ICUS and CCUS helps define patients at risk for hematologic evolution and informs monitoring intervals.
- These conditions blur traditional boundaries between “benign” unexplained cytopenia and hematologic malignancy.
- Their identification explains a subset of “unexplained” anemia in older adults that would otherwise be labeled idiopathic or “of aging.”
- Importantly, most patients remain stable and require no disease-directed therapy.
Spectrum and Overlap
- ICUS, CCUS, and MDS lie on a biologic and clinical continuum of clonal hematopoiesis.
- The key transitions are:
- Acquisition of clonality (ICUS → CCUS)
- Emergence of dysplasia (CCUS → MDS)
- This framework links back to the unexplained anemia of aging—many cases of “idiopathic” anemia may, in fact, represent early or low-level clonal hematopoiesis below current diagnostic thresholds.
- Conceptually, this closes the loop: the ABC spine explains mechanism, the Rule of Thirds defines population patterns, and the Clonal Spectrum maps molecular evolution.
In the Eye of the Beholder: What ICUS Really Means
The term idiopathic cytopenia of undetermined significance (ICUS) sounds precise but is, in practice, a label of uncertainty. It refers to persistent cytopenia without identifiable cause, mutation, or dysplasia and is often presented as the earliest point along a clonal spectrum leading toward CCUS and MDS. Yet this framing is largely in the eye of the beholder. Investigators defining the ICUS–CCUS–MDS continuum are studying a subset of patients with unexplained cytopenia through a molecular lens, not the broader population of patients with mild, stable, hypoproliferative anemia. For many such individuals, the explanation may lie outside the marrow entirely—subtle kidney dysfunction, low-grade inflammation, hormonal changes, or nutritional insufficiency.
ICUS, then, should not be equated with “unexplained anemia.” It represents one research-driven slice of that larger clinical territory, viewed through the narrow aperture of clonality. The distinction matters: not every patient whose anemia remains unexplained belongs on a clonal spectrum, and not every step on that spectrum reflects inevitable progression toward marrow failure.
Bringing It All Together: The Integrated Framework
Most clinicians approach normocytic, normochromic, hypoproliferative anemia through the A–B–C spine, a simple, practical algorithm that classifies causes into A: nutritional deficiency, B: inflammation, and C: organ dysfunction. It is the framework most of us use intuitively at the bedside, and for good reason: it is clear, teachable, and covers the majority of clinical scenarios.
Yet two additional worlds orbit this familiar structure. One is the world of aging, where the same mechanisms play out against the background of physiologic decline, creating a residual category of unexplained anemia of aging that exposes the limits of our current diagnostic resolution. The other is the world of clonality, where acquired somatic mutations blur the boundary between normal and malignant hematopoiesis. These pre-MDS states (ICUS, CCUS) expand our understanding of what a primary marrow process can mean, revealing molecular changes invisible to standard morphology or peripheral testing.
When viewed together, these three frameworks—clinical, epidemiologic, and molecular—form a layered model of hypoproliferative normocytic anemia. The A–B–C spine provides the functional map for clinical reasoning, the Rule of Thirds situates those mechanisms within population-level patterns, and the Clonal Spectrum illuminates one corner of organ dysfunction through a genetic lens. Together, they form a three-dimensional model that integrates physiology, epidemiology, and molecular pathogenesis. Each speaks a different dialect of the same language, and together they offer a more complete and nuanced understanding of this common yet complex form of anemia.
Returning to Mr. J’s case, we can now view his mild, stable normocytic anemia through this integrated lens. Through the A–B–C spine, nutritional deficiency (A), inflammation (B), and overt organ dysfunction (C) have all been reasonably excluded. From the standpoint of the Rule of Thirds, his anemia joins the substantial fraction of older adults whose evaluations remain unexplained, reminding us that his situation is common rather than exceptional. And viewed through the Clonal Spectrum, we can imagine the possibility, still largely theoretical in practice, that subtle age-related clonal hematopoiesis may contribute to his findings. In sum, this integrated view transforms what once seemed like a diagnostic dead end into a more nuanced understanding of how aging, regulation, and molecular drift converge in the marrow to produce a mild, persistent anemia.
How I explain this to a patient
Mr. J, your blood tests show a mild anemia that we can’t trace to any clear cause. I pause here for a beat; “we don’t know” can sound unsettling, and I want to give him space to react.
We’ve ruled out the usual possibilities: iron or vitamin deficiency, kidney or thyroid disease, inflammation, and any sign of a blood or bone cancer. In people your age, this situation is actually quite common; about a third of older adults with anemia fall into this “unexplained” group.
What that means is that the bone marrow simply isn’t making red cells at its usual pace, but we don’t see anything worrisome driving it. Sometimes I add that the machinery is slowing a little with age, not broken, just less efficient. It may reflect subtle hormonal or inflammatory changes that our current tests can’t yet detect.
We could do a bone-marrow biopsy, but it’s unlikely to change how we manage things. The important point is that we’re confident this isn’t leukemia or another cancer, and that your anemia is stable and mild. I make sure to end here, on reassurance and next steps.
We’ll keep monitoring your counts, focus on how you’re feeling, and revisit if anything changes.
In practical terms, this unified framework does not replace the A–B–C spine; it enriches it. Clinicians can still move step by step through nutritional, inflammatory, and organ-based causes, but now with added awareness that:
The goal is integration, not complication, to see hypoproliferative anemia as a spectrum rather than a set of silos. By situating the A–B–C spine at the hub of this unified framework, we preserve its clinical utility while extending its reach into the realities of aging and genomic hematology.
Figure 4. Integrating Mechanistic, Epidemiologic, and Clonal Frameworks. This synthesis unites the three complementary approaches to normocytic, normochromic, hypoproliferative anemia. The mechanistic (ABC) spine defines broad physiologic domains—nutritional, inflammatory, and organ-related. The epidemiologic (Rule of Thirds) lens shows how these categories distribute in aging populations. The clonal continuum (ICUS–CCUS–MDS) highlights marrow-level evolution and overlap with unexplained anemia. Seen together, these perspectives form an integrated conceptual map that links mechanism, population data, and pathobiology, underscoring that no single framework is sufficient in isolation.
Conclusion
Anemia remains one of the most familiar findings in medicine, yet its causes continue to unfold at every level of analysis. The challenge for clinicians is not simply to memorize frameworks, but to move fluidly among them, to think mechanistically at the bedside, epidemiologically in populations, and molecularly in the marrow. By approaching normocytic, normochromic anemia through these intersecting lenses, we preserve the clarity of traditional reasoning while remaining open to the deeper complexities that aging and genomics reveal.
Even with a structured algorithm such as the ABC spine, about 30 percent of normocytic anemias will remain unexplained after standard peripheral evaluation. Within that residual group lies a spectrum that ranges from subtle physiologic changes of aging to early pre-clonal states (ICUS, CCUS) that may evolve into overt marrow disease. The clinician’s task is to recognize this zone of uncertainty, to decide when observation is sufficient and when a bone marrow aspiration, biopsy, or molecular study may uncover early clonal evolution.
This balance between pattern recognition and biological humility is the essence of the approach. It allows us to translate frameworks into judgment at the bedside.
Test Your Understanding
Apply what you’ve learned with our interactive quiz on the approach to normocytic, hypoproliferative anemia.
Take the Quiz → - Definition and Features
- C1. Kidney