For Your Healthcare Provider
Have your patient scan this QR code with their smartphone camera to instantly access this educational guide on their device.

A guide for patients with hereditary spherocytosis (HS)
Access the Resources
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
Hereditary spherocytosis (HS) is a genetic condition in which red blood cells are shaped differently and break down more quickly than usual.
Most people with HS have a mild to moderate form, feel well day to day, and lead full, active lives with the right monitoring and care. HS can look different from person to person, and your doctor’s goal is to understand your pattern and support you over time.
This guide applies to outpatient care and does not replace emergency evaluation for rapidly worsening symptoms, severe jaundice in newborns, or high fever after splenectomy.
First things first
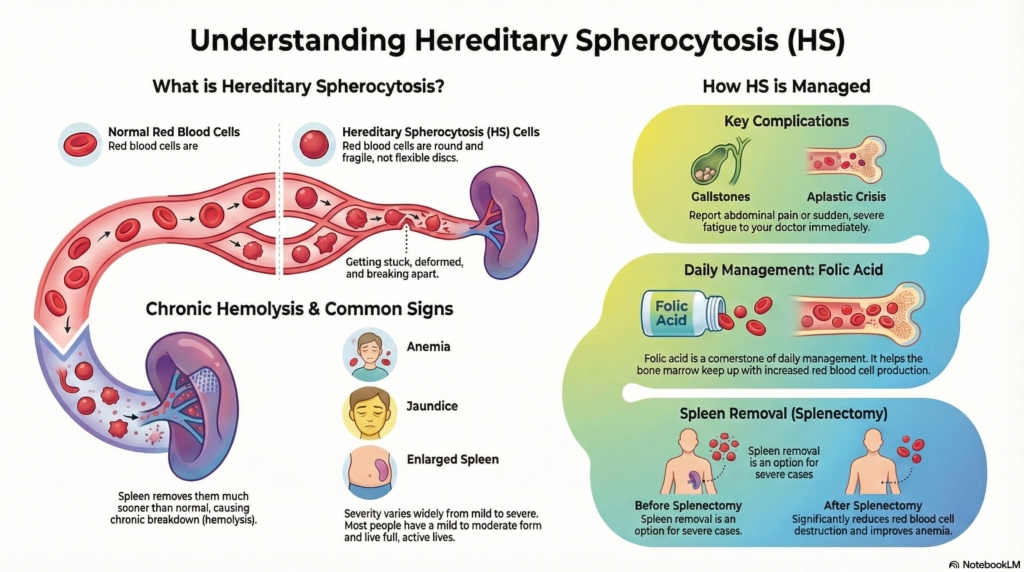
Hereditary spherocytosis is a lifelong red blood cell condition that can cause anemia, jaundice, and gallstones. Many people have mild disease and live normal, active lives, while others need closer monitoring or treatment. Understanding your pattern helps guide follow-up and long-term care.
What is hereditary spherocytosis?
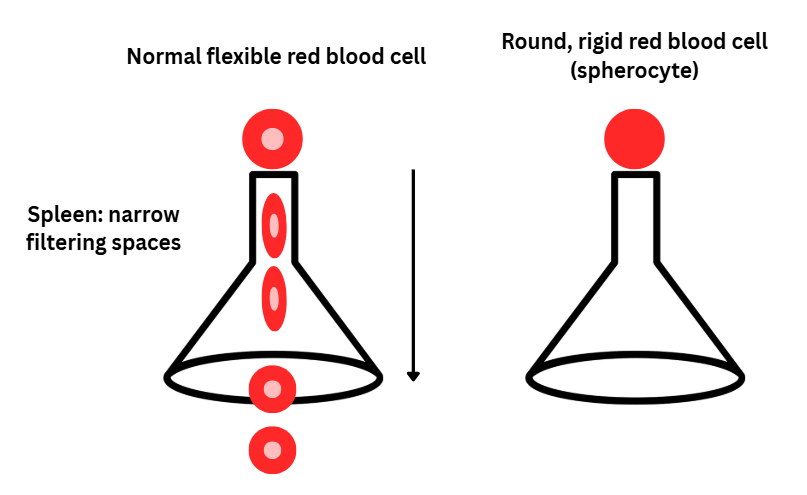
Hereditary spherocytosis is a genetic condition that affects the structure of the red blood cell membrane. Normal red blood cells are flexible discs that travel easily through narrow blood vessels. In HS, the cells become rounder (“spherocytes”) and less flexible, so they are removed more quickly by the spleen. This leads to ongoing red blood cell breakdown, called hemolysis.
HS often runs in families (usually autosomal dominant), though some cases occur for the first time in a person with no family history.
Severity varies:
- mild HS: minimal symptoms, normal or near-normal hemoglobin
- moderate HS: some anemia, more noticeable jaundice, risk of gallstones
- severe HS: significant anemia, need for transfusions, consideration of spleen removal
Why it happens (causes)
Genetic changes
HS results from changes in the genes that make proteins supporting the RBC membrane. These can include spectrin, ankyrin, band 3, or protein 4.2. Most people inherit HS in an autosomal dominant pattern, meaning a child has a 50% chance of inheriting it if one parent has HS. Some cases occur as new (de novo) mutations.
How spherocytes form
When membrane proteins are altered, RBCs lose their flexible, biconcave shape and become round. These spherocytes cannot pass easily through the spleen’s narrow channels, so they become trapped and are destroyed earlier than normal.
Does it cause symptoms?
Symptoms in newborns and infants (neonatal jaundice)
Some babies with HS develop jaundice in the first days of life. This may be more noticeable or last longer than typical newborn jaundice. Infants may also become anemic in the first months of life. Close monitoring helps prevent complications.
Symptoms in children and adults
Symptoms vary widely and may include:
- fatigue or low energy
- pale skin
- yellowing of the eyes or skin
- darker urine
- abdominal discomfort or early satiety from an enlarged spleen
- episodes of worsening anemia during illness
- abdominal pain, especially after eating fatty foods (possible gallstones)
- nausea or vomiting (possible gallstones)
Some people have very mild symptoms, while others need more frequent monitoring or treatment.
Is it dangerous?
Most people with HS do well with regular care, but some complications need attention.
Aplastic crisis
This occurs when the bone marrow temporarily stops making new red blood cells, often during certain viral infections. Symptoms include sudden fatigue, very pale skin, a fast heartbeat, or difficulty catching breath. It is treatable but requires urgent medical evaluation. This is most often triggered by parvovirus B19, especially in children, and leads to a rapid drop in new red blood cell production.
Hemolytic crisis
During infections, RBCs may break down faster than usual. This can lead to increased jaundice, darker urine, or worsening anemia. Hydration and medical follow-up help guide treatment.
Gallstones
Because RBCs break down more quickly, bilirubin levels rise over time, increasing the likelihood of pigment gallstones. These may cause abdominal pain, nausea, or vomiting and sometimes require surgery.
Problems from an enlarged spleen
The spleen may grow larger from working harder to remove abnormal RBCs. This may cause abdominal discomfort and, rarely, increase the risk of injury. Your doctor may advise avoiding certain high-impact activities.
How your doctor evaluates it
Evaluation usually includes:
- complete blood count (CBC)
- reticulocyte count
- bilirubin and other markers of hemolysis
- a blood smear showing spherocytes
- specialized tests that confirm red cell membrane fragility (such as EMA binding or osmotic fragility)
- abdominal ultrasound to check for gallstones
Family history often supports the diagnosis.
Genetic testing may be ordered but is not always required to diagnose hereditary spherocytosis. It may be used when:
- the diagnosis is unclear,
- symptoms begin very early in life,
- there is no known family history, or
- families want more detailed genetic information for future planning.
Most people are diagnosed based on their blood tests and clinical features alone.
How is it treated
Treatment depends on the severity of HS.
Folic acid
Because the bone marrow works harder to replace RBCs, folic acid supports increased red blood cell production. Many people take folic acid daily.
Splenectomy
Removing the spleen may be recommended for moderate to severe HS when anemia or symptoms significantly affect daily life. Splenectomy reduces red blood cell destruction and often improves energy and hemoglobin levels. After splenectomy, specific vaccines are required, and any fever is treated as an emergency.
Partial splenectomy
In some children, removing part of the spleen may reduce symptoms while preserving some immune function.
Treating gallstones
If gallstones cause pain or infection, the gallbladder may need to be removed. In some cases, gallbladder surgery is combined with splenectomy.
Daily life and self-care
Most people with HS live normal, active lives. Take folic acid consistently, attend regular follow-up visits, stay hydrated, and report new symptoms such as increasing fatigue, jaundice, or abdominal pain. If you have had a splenectomy, fever is always an urgent concern and should be evaluated promptly.
When should I contact my doctor?
Contact your doctor if you notice:
- worsening fatigue or paleness
- increasing yellowing of the skin or eyes
- abdominal pain (possible gallstones or spleen issues)
- very dark urine
- fever if you do not have a spleen
Seek urgent care for:
- severe weakness
- trouble breathing
- fainting
- sudden abdominal pain
What is the usual plan going forward?
HS is lifelong but manageable. Many people have mild to moderate symptoms and need regular monitoring, folic acid, and occasional evaluations for anemia or gallstones. Some people benefit from splenectomy if symptoms remain significant. Long-term follow-up helps prevent complications.
Making sense of it
Think of red blood cells as flexible disks that easily bend to travel through tiny pathways.
In HS, the cells become round and less flexible, so the spleen removes them sooner than expected.
Key takeaways
- HS makes red cells round and fragile, causing them to break down sooner
- most cases are mild to moderate, and many people live active lives with routine care
- anemia and jaundice are common, especially during illness
- gallstones can occur, so abdominal pain should be reported
- splenectomy is an option when symptoms are more severe
- regular follow-up is important, especially during childhood and after infections
For clinicians: Read our detailed guide on how to communicate about HS to patients.