Why cold agglutinin disease is best understood as a complement-driven hemolytic anemia
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
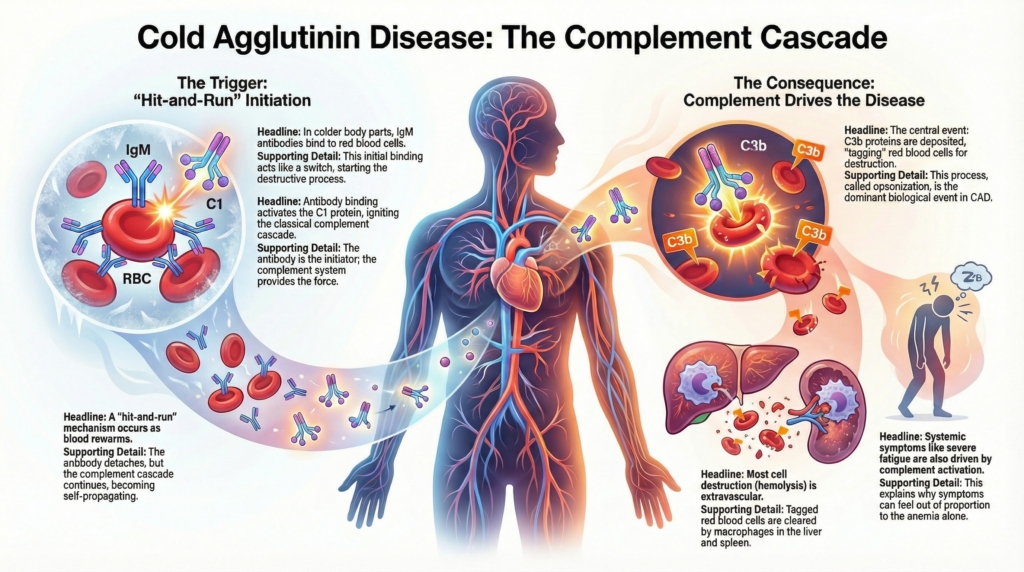
Cold agglutinin disease is best understood as a classical complement pathway–driven hemolytic disorder. In CAD, antibody binding initiates the process, but complement activation determines much of the phenotype: the degree of hemolysis, the pattern of red-cell clearance, and the logic of effective therapy.1
Understanding CAD therefore requires shifting attention away from antibody binding alone and toward the kinetics, amplification, regulation, and consequences of complement activation, especially at the level of C1 and C3. This helps explain why CAD behaves differently from warm autoimmune hemolytic anemia, why corticosteroids are usually ineffective, and why proximal complement inhibition can be highly effective.2
This spoke explains how complement biology shapes hemolysis, symptoms, and therapeutic vulnerability in CAD. Before turning to CAD specifically, it is useful to orient the complement system around one central idea: different pathways enter the cascade in different ways, but they converge on C3.
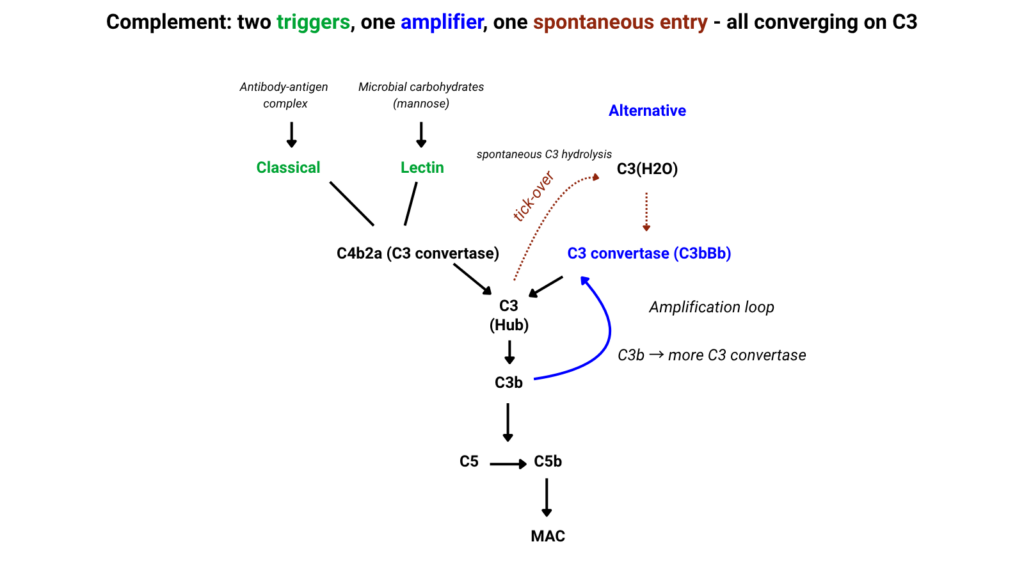
For CAD, this map should be read selectively. The disease is initiated primarily through IgM-mediated classical pathway activation, not through tick-over or lectin pathway recognition. The alternative pathway matters mainly because deposited C3b can feed an amplification loop, extending the consequences of an initiating antibody event.
The initiating event: IgM and C1 engagement
In primary CAD, the pathogenic antibody is almost always IgM.3
IgM is far more efficient at complement activation than IgG because a single pentameric IgM molecule presents multiple Fc regions in close proximity, allowing strong C1q engagement. In contrast, IgG molecules must cluster densely on the red-cell surface to achieve comparable activation. This structural difference explains why relatively small amounts of IgM can trigger robust complement activation.4
When bound to red blood cells, pentameric IgM undergoes a conformational change that exposes binding sites for C1q, triggering complement activation even at relatively low antibody densities.5
Key features:6
- IgM binds red cells most efficiently at lower temperatures
- C1q binding activates C1r and C1s
- Complement activation proceeds rapidly once initiated and may continue after antibody disengagement, as complement components remain bound to the red cell surface
- Antibody quantity matters less than thermal amplitude and complement-fixing efficiency
The antibody acts as a switch. Complement supplies the force.7
Classical pathway dominance in CAD
In CAD, complement activation is initiated through, and largely dominated by, the classical pathway.8
This has several consequences:9
- alternative and lectin pathways are not primary drivers
- C1 activation is the critical upstream event
- downstream amplification depends on intact C3 convertase formation
Once C1s is activated, it cleaves C4 and C2, forming the classical C3 convertase (C4b2a). This step commits the system to amplification..10
At this point, complement activation can continue and amplify even after the initial antibody-binding event has passed.11
Because C4 is consumed early in classical pathway activation, patients with active CAD may show lower C4 levels relative to C3, a pattern that supports ongoing classical-pathway engagement rather than generalized complement depletion.12
C3 as the central effector molecule
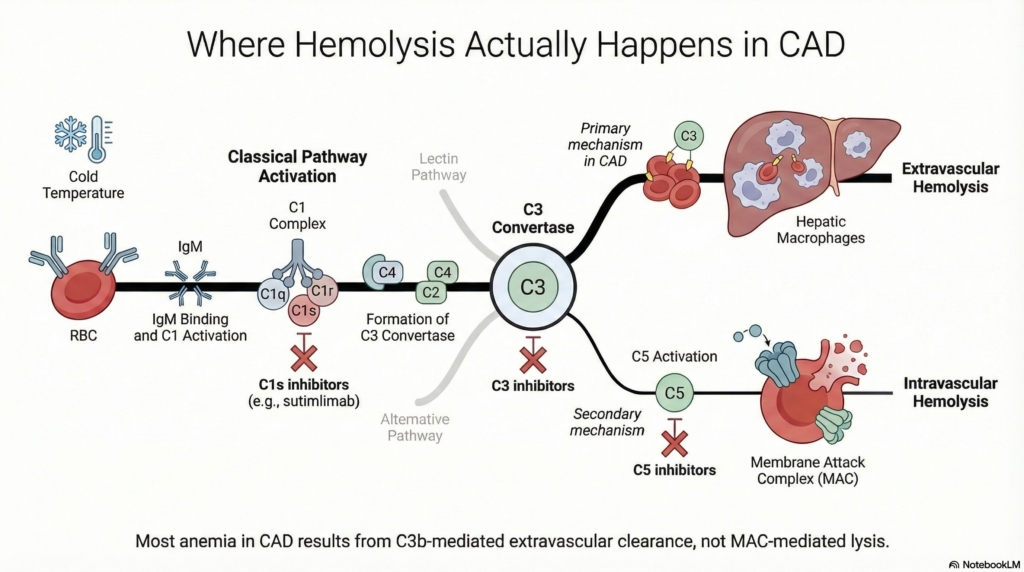
The dominant biologic event in CAD is C3 activation and deposition, not terminal complement lysis.13
After C3 convertase formation:14
- C3 is cleaved into C3a and C3b
- C3b covalently deposits on the red cell surface
- red cells become opsonized for clearance
Most hemolysis in CAD is therefore:15
- extravascular, mediated by macrophages in the liver
- driven by C3b and iC3b recognition
- largely independent of membrane attack complex formation
This explains why:16
- hemolysis can be chronic and compensated
- LDH and bilirubin elevations may be modest
- haptoglobin may be low but not absent
CAD is a disease of opsonization, not explosive lysis.
Surface-bound C3b is progressively cleaved during circulation to iC3b and then to C3d. Earlier fragments promote macrophage recognition and clearance, whereas C3d represents a more stable remnant that persists on surviving cells. This processing explains why the DAT in CAD typically detects C3d rather than C3b and why complement positivity may remain even when hemolysis is clinically improving.17
Once initial C3b is deposited through classical pathway activation, it can recruit factor B and factor D to form an alternative pathway amplification loop, generating additional C3 convertase and further opsonization. Thus, transient antibody binding can initiate a cascade that amplifies independently of continued classical pathway signaling.18
The sequence is therefore: antibody binding initiates, complement amplifies, and C3-mediated opsonization drives clearance.
The “hit-and-run” nature of complement in CAD
A defining feature of CAD is that complement activation often continues after antibody disengagement.19
This occurs because:20
- IgM binds transiently at cooler peripheral temperatures
- complement activation persists as blood returns to central circulation
- C3b remains fixed to the red cell surface
At this point, the key question is: is ongoing hemolysis being driven by continued antibody binding, or by complement that has already been set in motion?
The answer is that complement, once activated, can sustain the process:21
- hemolysis can continue after rewarming
- disease activity may persist despite cold avoidance
- symptoms do not correlate tightly with measured antibody binding
Complement activation has memory. The antibody does not need to remain bound.
Why intravascular hemolysis is limited
Although terminal complement activation can occur, MAC-mediated intravascular hemolysis is usually limited in CAD.22
Reasons include:23
- efficient regulation of terminal complement on red cells
- rapid clearance of opsonized cells before MAC formation
- predominant C3-mediated extravascular removal
However::24
- severe disease or high complement activity can overwhelm regulation
- intravascular hemolysis may occur during acute exacerbations
- complement inhibition upstream prevents both pathways
This balance explains variability in laboratory severity across patients.
Red cells are normally protected from terminal complement injury by membrane regulatory proteins, especially CD55 (decay-accelerating factor) and CD59, which respectively disrupt convertase formation and prevent membrane attack complex assembly. Because these regulators are intact in CAD, terminal complement activation is usually contained before intravascular lysis occurs. Only when complement activation is unusually intense can regulatory capacity be exceeded, allowing intravascular hemolysis to emerge.25
Complement and symptoms beyond anemia
Complement activation likely contributes to symptoms that are not explained by hemoglobin alone.26
Potential mechanisms include:
- release of anaphylatoxins (C3a, C5a)
- endothelial activation
- microvascular dysfunction
- inflammation and fatigue
These effects help explain:27
- fatigue out of proportion to anemia
- cold-induced pain and circulatory symptoms
- patient-reported burden exceeding laboratory expectations
CAD is not only a disorder of red-cell clearance; it is a complement-driven disease whose clinical burden may exceed what is captured by the hemoglobin level.
Why corticosteroids are usually ineffective
Corticosteroids are usually ineffective in CAD because they do not meaningfully interrupt the dominant complement-mediated mechanism of hemolysis.28
Corticosteroids:
- suppress Fc-mediated immune clearance
- reduce IgG-driven inflammation
- have little impact on complement activation
In CAD:29
- hemolysis is complement-driven
- antibody production is clonal and steroid-resistant
- macrophage clearance is complement-opsonin mediated
Steroids do not meaningfully interrupt the disease pathway.
Therapeutic implications of complement biology
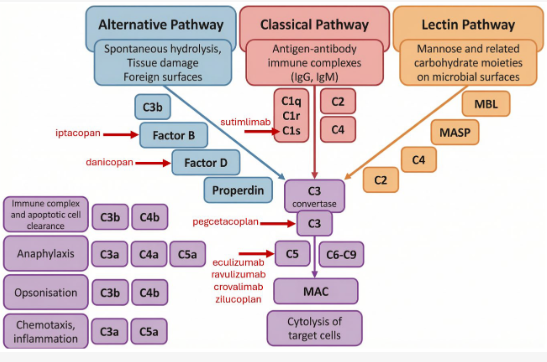
Complement biology explains the success of proximal complement inhibition in CAD.
Targeting C1s:30
- blocks classical pathway initiation
- prevents downstream C3 deposition
- halts hemolysis without broad immunosuppression
This approach:
- acts upstream of amplification
- preserves alternative pathway function
- aligns precisely with CAD pathophysiology
Complement inhibition works not because CAD is severe, but because it is mechanistically clean.
Explicit principle
In CAD, the antibody initiates disease, but complement determines phenotype.31
Severity, chronicity, symptoms, and treatment response are shaped less by how much antibody is present than by how efficiently complement is activated and sustained.32
Understanding complement biology is therefore not optional. It is the key to understanding why CAD behaves the way it does — and why it responds to the therapies that work.
Reflect and Apply
A patient with CAD has low C4 and modestly low C3.
Explain this pattern mechanistically.
Test your thinking
A short, judgment-focused quiz on complement biology in cold agglutinin disease.