How clone, antibody, temperature, and complement converge
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
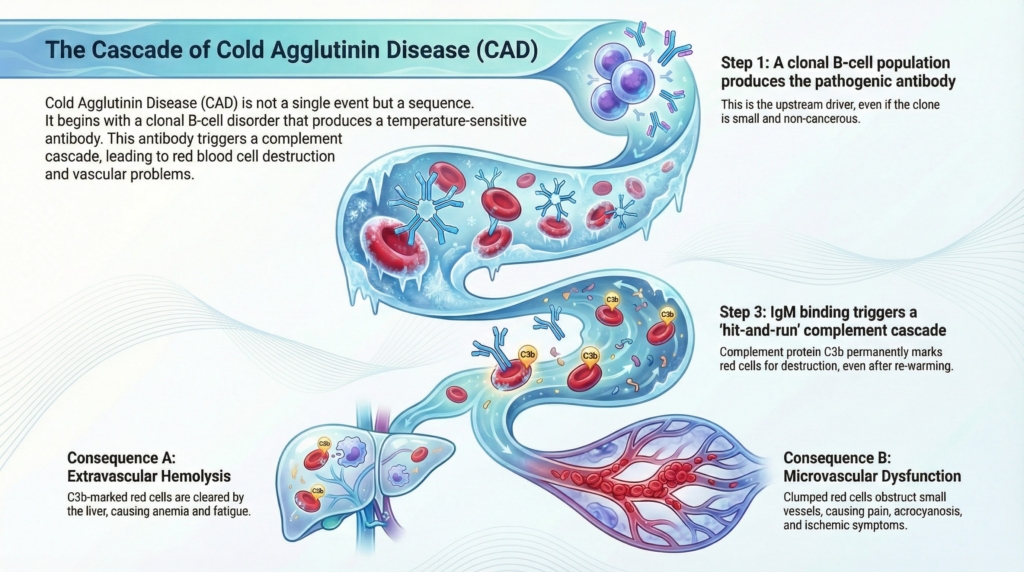
Cold agglutinin disease (CAD) is a pathophysiologic cascade: each downstream event can only be understood in light of what precedes it. A clonal B-cell process gives rise to a temperature-sensitive antibody, which binds red blood cells in the cold, activates complement, and drives injury through hemolysis and vascular dysfunction. Following this cascade in order is essential, because downstream manifestations cannot be interpreted without understanding what precedes them.1
The clonal B-cell disorder: the upstream driver
CAD is fundamentally linked to a monoclonal B-cell lymphoproliferative process that is usually low-grade and clinically indolent.2 In many patients, the clone exists below the threshold that would otherwise be labeled lymphoma.3 Nevertheless, its biologic consequences are decisive, because it produces the pathogenic antibody that drives everything downstream.4
Key features of the clonal disorder:5
- most patients harbor a small clonal B-cell population in the bone marrow
- lymphoplasmacytic or marginal-zone–like phenotypes are most common
- overt lymphoma is uncommon at presentation
- clinical severity and symptom burden correlate poorly with clone size or marrow involvement
- clone-directed therapy reduces antibody production but does not immediately halt complement already activated
This explains why CAD behaves more like an immune-mediated disorder than a conventional lymphoma, despite its clonal origin.6
The pathogenic antibody: IgM with thermal amplitude
The hallmark of CAD is a monoclonal IgM antibody directed against red blood cell surface antigens, most commonly I or i.7 What makes this antibody pathogenic is not simply its specificity, but its thermal amplitude, the highest temperature at which it binds red cells.8
Key properties of the pathogenic IgM antibody:9
- IgM binds red cells most avidly at lower temperatures
- binding occurs primarily in cooler peripheral circulation
- pathogenicity depends more on thermal amplitude than antibody titer
- antibodies active at warmer temperatures produce more severe disease
A low-titer antibody active at 32–34°C may cause more severe disease than a high-titer antibody that binds only at very low temperatures, explaining why antibody quantity alone is misleading.10
Once IgM binds in cooler peripheral circulation, it can rapidly fix complement. This can commit the red cell to complement-mediated injury even after IgM dissociates during rewarming. The key determinant is therefore not simply whether antibody is present, but where binding occurs, at what temperature, and how efficiently complement is fixed.11
Complement activation: the central amplifier
Complement is the dominant effector in CAD. IgM binding efficiently activates the classical complement pathway, and once initiated, this cascade determines both hemolysis and many clinical manifestations.12
Key complement-mediated events:13
- C1 activation occurs immediately upon IgM binding
- C3b deposition marks red cells for clearance
- hemolysis is predominantly extravascular via hepatic macrophages
- intravascular hemolysis is usually limited but can occur during intense activation
- complement activity may continue after red cells return to central circulation
This “hit-and-run” mechanism explains why symptoms persist after cold exposure ends and why complement inhibition is therapeutically effective.14
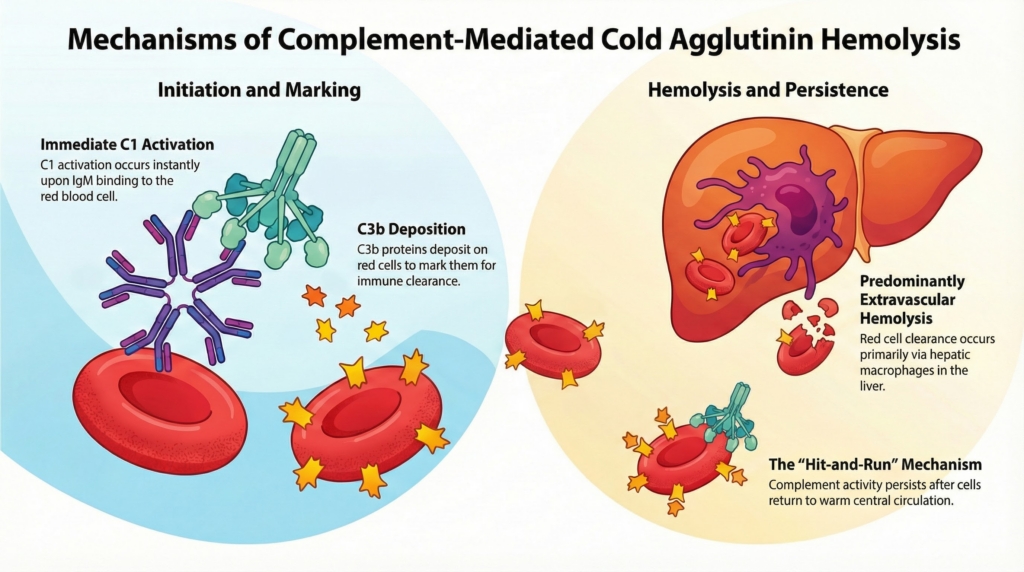
Hemolysis in CAD: mostly C3-mediated, mostly extravascular
Although cold agglutinin disease is often described simply as a hemolytic anemia, the mechanism of hemolysis is distinctive. Red cell destruction in CAD is driven primarily by classical complement pathway activation, C3b deposition, and hepatic macrophage clearance, rather than by Fc-mediated splenic clearance or widespread terminal complement lysis. Both extravascular and, less commonly, intravascular hemolysis can occur, but most anemia reflects C3-mediated extravascular clearance.15
Once IgM binds in cooler peripheral circulation, it activates the classical complement pathway and leads to deposition of C3b on the red-cell surface. Red cells heavily opsonized with C3b are recognized and removed predominantly by macrophages in the liver, producing extravascular hemolysis. This hepatic pattern is one of the key biologic differences between CAD and warm autoimmune hemolytic anemia, where splenic Fc-mediated clearance is usually more prominent.
Complement activation, however, does not have the same outcome on every red cell. In some cells, complement activation is incomplete or self-limited: surface-bound C3b is degraded to C3d, which is no longer efficiently recognized by hepatic macrophage receptors. These C3d-coated red cells may therefore remain in circulation despite evidence of complement activation. This balance between clearance and survival helps explain why anemia in CAD is often moderate rather than profound.
Key features of hemolysis in CAD:16
- C3b-opsonized red cells are cleared primarily in the liver
- anemia is often moderate rather than profound
- LDH and bilirubin may be only modestly elevated
- reticulocyte responses are variable17
- hemoglobin correlates poorly with fatigue and functional impairment
This explains the hemolytic component of CAD, but not the entire clinical phenotype. Hemoglobin, LDH, bilirubin, and reticulocyte count describe red-cell destruction; they do not fully capture cold-induced circulatory symptoms, pain, fatigue, or functional limitation. Those symptoms require a second layer of explanation: the mechanical and rheologic effects of IgM-mediated red-cell agglutination in cooler vascular beds.18
Agglutination and microvascular dysfunction: beyond hemolysis
At temperatures commonly reached in acral circulation, often below 30°C, pentameric IgM can bridge adjacent red cells, forming reversible aggregates that increase blood viscosity and impair laminar flow in small vessels. In digital arteries and capillary beds, this promotes functional stasis, reduced oxygen delivery, and ischemic discomfort, even in the absence of significant hemolysis.19
At temperatures commonly reached in acral circulation (often below 30 °C), pentameric IgM can bridge adjacent red cells, forming reversible aggregates that increase blood viscosity and impair laminar flow in small vessels. In digital arteries and capillary beds, this promotes functional stasis, reduced oxygen delivery, and ischemic discomfort even in the absence of significant hemolysis.
Key vascular effects:20
- impaired microcirculatory flow
- acrocyanosis, livedo, and cold-induced pain
- symptoms may occur without significant hemolysis
- ischemic discomfort may dominate the clinical picture
Complement, inflammation, and thrombosis
CAD is associated with an increased risk of thrombosis, a clinical observation that has been consistently reported across cohorts and practice settings. While the precise mechanisms remain an area of active investigation, this risk appears to arise from systemic inflammation and vascular perturbation linked to complement activation rather than from a classic inherited or acquired hypercoagulable state.21
Several interacting processes are thought to contribute:22
- activation of the classical complement pathway generates inflammatory complement fragments that can promote endothelial activation
- ongoing hemolysis contributes to systemic inflammation through release of free hemoglobin and downstream immune signaling
- IgM-mediated red cell agglutination in cooler vascular beds may produce localized microvascular stasis and endothelial stress
- thrombotic risk appears higher during periods of active disease and increased cold exposure, suggesting interaction between immune activity and environmental triggers
Taken together, these observations reinforce that CAD is better viewed as a complement-mediated hemolytic and circulatory disorder with potential systemic vascular consequences, rather than an isolated hemolytic anemia.23
Why pathophysiology matters clinically
Each layer of cold agglutinin disease pathophysiology maps to a distinct therapeutic and counseling implication. Treating CAD effectively requires recognizing which part of the cascade is driving a given patient’s symptoms, because no single intervention addresses all components of the disease.24 The task is not to ask “how low is the hemoglobin?” but “which part of the cascade is dominant in this patient?
Different interventions act at different levels of the cascade:25
- clone-directed therapy reduces production of the pathogenic IgM antibody but does not immediately reverse complement already fixed on circulating red cells
- complement inhibition interrupts ongoing hemolysis and inflammation, regardless of clone size or antibody titer
- warming strategies and cold avoidance reduce IgM binding and agglutination, directly improving vascular symptoms
- symptom severity often reflects environmental exposure and complement activity more than bone marrow disease burden
Understanding where a patient’s dominant burden lies helps avoid mismatched expectations, such as treating the clone when symptoms are driven primarily by complement-mediated hemolysis, or focusing on hemoglobin alone when vascular symptoms predominate.26
Key takeaways
- clonal origin: CAD begins with a monoclonal B-cell disorder but behaves as an immune-mediated disease
- thermal amplitude: IgM thermal amplitude, not antibody quantity alone, determines severity
- complement amplification: complement activation is the central amplifier of injury
- beyond hemolysis: hemolysis explains anemia, but not the full symptom burden
- vascular morbidity: vascular dysfunction and inflammation are major contributors to morbidity
Cold agglutinin disease is best understood not as a single mechanism, but as a sequence. Following that sequence clarifies why the disease looks the way it does — and why no single intervention is sufficient on its own.
Reflect and Apply
When evaluating a patient with cold agglutinin disease, try to locate the dominant problem within the cascade.
Is the clinical picture driven mainly by complement-mediated hemolysis, cold-induced red cell agglutination and microvascular symptoms, or both?
How would that distinction change what you monitor, what you explain to the patient, and what type of treatment you would expect to help most?
Test your thinking
A short, judgment-focused quiz on pathophysiology of cold agglutinin disease.