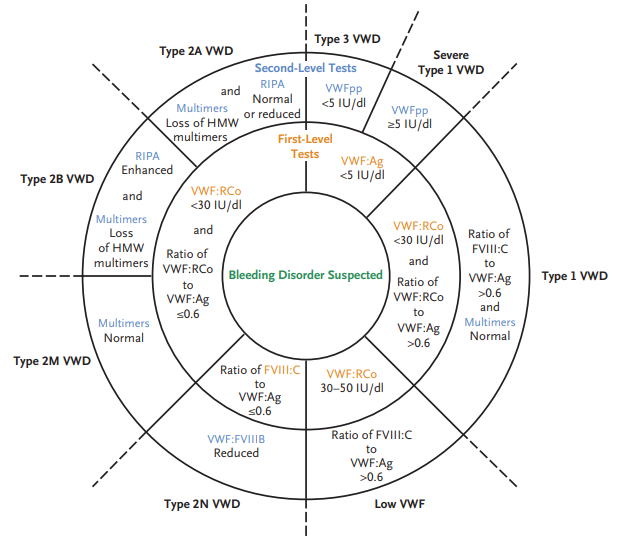
Type 2 VWD is characterized by a qualitative abnormalities in the VWF protein. Different subtypes reflect which protein-protein interactions are affected. In some cases, reduced binding to a physiologic binding partner may be caused by defective multimerization rather than a defect in a specific protein binding domain:
- Accounts for about 20% of cases of vWD.
- Autosomal dominant inheritance.
- Subtypes include:
- Type 2A:
- Most common subtype.
- Accounts for 10 to 15 percent of VWD cases.
- Deficiency of large (intermediate and high-molecular-weight) multimers due to either:
- Decreased multimer assembly
- Increased proteolysis
- Impaired von Willebrand factor (vWF) binding to collagen and platelets.
- Type 2B:
- Accounts for approximately 5 percent of VWD cases.
- Gain-of-function mutation which causes increased vWF binding to platelet GPIb alpha with rapid clearance of platelet-vWF complex.
- Enhanced binding tom platelets leads to accelerated clearance or sequestration of platelets and of the bound HMW VWF multimers, resulting in deficiency of high-molecular-weight multimers and thrombocytopenia (the latter occurring in 40% of affected patients).
- Phenocopy of platelet type vWD.
- Type 2M (M for multimer):
- Loss-of-function mutation which decreases vWF binding to platelet GPIb alpha or collagen.
- Normal vWF multimer distribution.
- Type 2N (N for Normandy, the location where it was discovered):
- Loss-of-function mutations in vWF that cause decreased vWF binding to factor VIII with abnormally increased clearance of factor VIII.
- May mimic mild hemophilia A.
- Type 2A:
- Most cases caused by missense mutations, which are usually limited to specific functional domains.
- Ratio of von Willebrand factor ristocetin cofactor activity to von Willebrand factor antigen (vWF:RCo to vWF:Ag) typically < 0.7, with exception of type 2N and collagen-binding variant of type 2M.
Learn more here.