How a laboratory curiosity became a complement-mediated clonal B-cell disorder
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
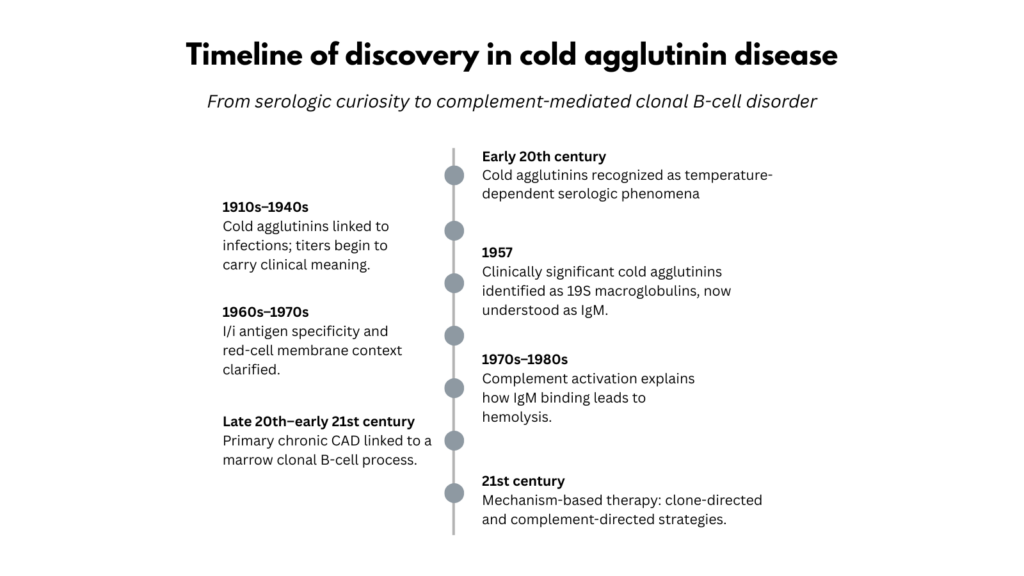
The history of cold agglutinin disease illustrates how a clinical disorder can emerge at the intersection of several scientific traditions: first appearing as a serologic curiosity, later explained through immunohematology and complement biology, and ultimately recognized as a complement-mediated hemolytic anemia linked to clonal B-cell disease.
Early 20th century — A temperature-dependent curiosity
Cold agglutinins were initially recognized as a serologic phenomenon rather than as a distinct clinical disease. In agglutination-based laboratory work, investigators observed that some sera could cause red cells to clump more strongly at lower temperatures and lose reactivity on warming, a property that complicated interpretation of serologic testing.1
At the time, the phenomenon lived largely in the world of methods rather than medicine. Different investigators described “autohemagglutinins,” “cold hemagglutinins,” and related cold-reactive antibodies without a unified concept of a temperature-dependent immune process affecting human red cells.
Clinicians had already reported occasional cases of autohemagglutination and cold-triggered vascular symptoms, but these remained isolated observations rather than a recognized syndrome.
In retrospect, this pattern is familiar in hematology: phenomena first dismissed as technical artifacts often become windows into deeper biological mechanisms.
1910s–1940s — From laboratory artifact to infection marker
Over the next several decades, physicians began noticing that cold agglutinins appeared in patients with certain illnesses, particularly respiratory infections.2
By the early 1940s, systematic studies showed that low-titer cold agglutinins were frequently observed and often transient. They appeared in a range of acute and chronic illnesses, including atypical pneumonia, other respiratory infections, and infectious mononucleosis.3
These observations helped establish cold agglutinins as clinically meaningful antibodies, but they did not yet define primary chronic cold agglutinin disease. Many infection-associated cold agglutinins were transient; primary CAD would later be understood as a persistent hemolytic disorder driven by a monoclonal IgM-producing B-cell clone.4
Importantly, these studies also introduced quantitative thinking. Low titers were common and nonspecific, whereas high titers were rare and strongly associated with severe atypical pneumonia.5 Even at that time, investigators recognized that the clinical meaning of cold agglutinins depended not simply on their presence but on their magnitude and context.6
Despite these observations, the pathogenic significance of cold agglutinins remained uncertain. Editorial commentary of the era emphasized that cold agglutinins were “usually harmless,” noting that they were inactive at body temperature and therefore unlikely to cause hemolysis under normal circumstances.7
In retrospect, this period represents a stage in which clinicians learned to separate signal from noise, but stopped short of recognizing primary chronic CAD as a distinct hemolytic disorder. In part this was because chronic anemia accompanied by cold sensitivity was rarely framed as a unified clinical entity, and in part because many observed cold agglutinins were transient infection-associated antibodies rather than markers of a persistent clonal process.8
1957 — The macromolecular nature of cold agglutinins
A major conceptual advance came in the late 1950s when Fudenberg and Kunkel demonstrated that clinically significant cold agglutinins were 19S macroglobulins, the immunoglobulin class now known as IgM.9
Using zone electrophoresis and ultracentrifugation, they showed that cold agglutinin activity consistently sedimented with the 19S fraction, whereas the antibodies responsible for warm autoimmune hemolytic anemia resided in the 7S fraction (IgG). This distinction helped establish that cold and warm autoimmune hemolytic anemias were immunologically distinct disorders.10
Their clinical material also hinted at another important insight. Some patients with chronic cold agglutinin disease showed features suggestive of an underlying lymphoproliferative process, including lymphadenopathy, marrow lymphoid infiltration, and circulating macroglobulins.11 Although the significance of this observation was not yet fully appreciated, .it foreshadowed the later recognition that cold agglutinin disease often arises from a clonal B-cell process.12
Although these observations hinted at a clonal origin, the tools required to characterize marrow lymphoproliferative disorders did not yet exist, and cold agglutinin disease continued for decades to be classified primarily as a form of autoimmune hemolytic anemia.13
This work marked a turning point. Cold agglutinins were no longer simply unusual antibodies. They were defined immunoglobulins with a specific molecular identity, and in modern terms this was the moment when cold agglutinin disease could no longer be treated as merely a variant of warm autoimmune hemolytic anemia.14
1960s–1970s — Antigen specificity and membrane biology
The next stage in the story came when immunohematologists began to dissect what these IgM antibodies recognized on red cells.
Investigators defined the serologic importance of the I/i antigen system, whose determinants are carbohydrate structures carried on red-cell membrane glycoproteins and glycolipids. Anti-I antibodies reacted more strongly with adult red cells, whereas anti-i antibodies preferentially reacted with cord cells. This helped explain why clinically significant cold agglutinins in adults almost always display anti-I specificity and why cord blood behaves differently in serologic testing.15
Experiments by Rosse and colleagues added a crucial mechanistic insight. They demonstrated that when the I antigen was extracted from the red-cell membrane and placed in solution, it reacted with antibody equally well at 37 °C and 0 °C. In the intact membrane, however, antibody binding was strongly temperature dependent.16
This observation suggested that the apparent“cold specificity” of these antibodies was not solely an intrinsic property of the immunoglobulin, but also reflected how the I antigen was presented within the red-cell membrane environment.17
This work placed cold agglutinins squarely within the emerging field of immunohematology, transforming nonspecific clumping phenomena into antigen-specific IgM antibodies interacting with defined membrane structures.18
1970s–1980s — Complement biology connects agglutination to hemolysis
Even after antigen specificity was established, an important question remained: how does cold-reactive IgM binding lead to hemolysis?
Earlier investigators had proposed explanations such as increased mechanical fragility of clumped red cells. Mechanistic studies in the late twentieth century replaced these ideas with a clearer model based on complement activation.
Mechanistic studies showed that when IgM cold agglutinins bind to red cells at lower temperatures, they efficiently fix the first component of complement (C1), initiating the classical complement pathway. This process leads to deposition of complement fragments, particularly C3, on the red-cell membrane.19
Work by Rosse and Adams demonstrated that the severity of hemolysis reflects a combination of interacting variables: the concentration of antibody present, the thermal amplitude (the highest temperature at which the antibody can bind), the efficiency with which the antibody fixes complement, and the extent to which complement activation is regulated on the red-cell surface.20
These studies also clarified why clinical manifestations vary widely among patients. Some antibodies produce strong agglutination but limited complement activation, leading to acrocyanosis with little anemia. Others efficiently activate complement, producing chronic hemolysis. Most hemolysis occurs through extravascular clearance of C3-coated red cells in the liver, whereas intravascular hemolysis may occur when complement activation proceeds further along the cascade.21
This era transformed cold agglutinin disease conceptually. What had once been viewed as temperature-dependent clumping was now understood as classical-pathway complement–mediated hemolysis with measurable determinants of disease severity.22
Late 20th century to early 21st century — Recognition of a clonal B-cell disorder
For many years, primary chronic cold agglutinin disease was classified largely as a form of autoimmune hemolytic anemia of unclear origin.23
Gradually, however, clinical and pathologic studies revealed that most patients with primary chronic CAD produce a monoclonal IgM antibody, often κ-restricted, derived from a clonal B-cell population.24
Bone marrow studies eventually demonstrated a characteristic lymphoproliferative disorder composed of small monoclonal B cells forming nodular aggregates in the marrow. These cells display a distinctive immunophenotype and frequently use the IGHV4-34 gene segment, which encodes part of the antibody’s antigen-binding region and is strongly associated with antibodies that recognize the I antigen on red cells.2527
These findings reframed the disease. Rather than a purely autoimmune phenomenon, primary cold agglutinin disease came to be recognized as a complement-mediated hemolytic anemia driven by a marrow clonal B-cell lymphoproliferative disorder that produces a pathogenic monoclonal IgM autoantibody.28
21st century — Mechanism-based therapy
After a century of discovery, these mechanistic insights eventually translated into targeted therapy. Once complement activation and clonal B-cell production of IgM were established as the twin pillars of disease pathogenesis, each became an explicit therapeutic target..29
Two mechanistic treatment strategies emerged.
Clone-directed therapies, such as rituximab-based regimens, aim to reduce the B-cell population producing the pathogenic IgM antibody. These treatments may produce durable remissions but typically act slowly and carry risks of immunosuppression.30
Complement-directed therapies, particularly proximal classical-pathway inhibition, can rapidly reduce complement-mediated hemolysis, but they do not eliminate the underlying IgM-producing clone and usually require ongoing administration to maintain benefit.31
Complement inhibition can improve hemolysis and anemia, whereas cold-induced circulatory symptoms may persist because they are driven primarily by red-cell agglutination rather than complement activation.32
Thus modern treatment reflects the dual biology of the disease: one axis targeting the clone, the other targeting the complement pathway.
The conceptual arc of the field
Seen across more than a century, the history of cold agglutinin disease reflects the convergence of several scientific traditions.
Immunohematology transformed cold-reactive autohemagglutinins from laboratory curiosities into antigen-specific IgM antibodies directed against defined red-cell membrane structures.
Complement biology revealed that hemolysis results from classical-pathway activation triggered by IgM bound at low temperatures, with quantifiable determinants such as thermal amplitude and complement fixation efficiency.
Clonal hematology showed that, in primary chronic CAD, the pathogenic antibody usually arises from a stereotyped B-cell population in the bone marrow.
These insights did not emerge in a simple linear sequence. Each was glimpsed partially before the others were fully understood. Together they transformed cold agglutinin disease from a puzzling serologic anomaly into a clinical disorder that sits at the intersection of immunology, complement biology, and clonal hematology.
For clinicians and trainees, this arc is a reminder that diseases located at the boundary between autoimmunity, complement biology, and clonal hematology rarely fit neatly into a single category. The history of cold agglutinin disease shows how classification, mechanism, and therapy evolve together over time, often slowly and nonlinearly, as new scientific tools reveal dimensions of disease that earlier observers could only partially glimpse.
Reflect and Apply
The history of cold agglutinin disease shows how a disorder can move from laboratory curiosity to mechanistic disease as new tools become available.
Pause and ask:
What was the key conceptual shift: recognizing the antibody, recognizing complement, or recognizing the clone?
How did each discovery change what clinicians could explain, diagnose, or treat?
Where in modern hematology might we still be mistaking a technical artifact for an early clue?
The goal is not to memorize dates. The goal is to see how classification, mechanism, and therapy evolve together.
Test your thinking
A short, judgment-focused quiz on the history of cold agglutinin disease.
Additional Reading
Early clinical observations and interpretation:
- Favour CB. Autohemagglutinins. I. Cold agglutinins. J Clin Invest. 1944;23(6):890-897.
- Anonymous (editorial). The cold agglutinins. Br Med J. 1944;2(4350):16–17. (Embedded in: “The cold agglutinins” editorial section of the British Medical Journal.)
Immunoglobulin characterization:
- Fudenberg HH, Kunkel HG. Physical properties of the red cell agglutinins in acquired hemolytic anemia. J Exp Med. 1957;106(5):689-702.
Antigen specificity and membrane biology:
- Rosse WF, Lauf PK. Reaction of cold agglutinins with I antigen solubilized from human red cells. Blood. 1970;36(6):777-784.
- (Within Rosse & Adams and Rosse & Lauf as cited above): Rosse WF, Sherwood JB. Cold reacting antibodies: differences in the reaction of anti‑I antibodies with adult and cord red blood cells. Blood. 1970;36:28–42.
Complement activation and clinical variability:
- Schreiber AD, McDermott P, Rosse WF, et al. Low-titer cold-hemagglutinin disease. N Engl J Med. 1977;296:1490–1494. Followed by correspondence: Abramson N. Cold agglutinins. N Engl J Med. 1977;297:727–728; reply by Schreiber AD. N Engl J Med. 1977;297:728.
- Rosse WF, Adams JP. The variability of hemolysis in the cold agglutinin syndrome. Blood. 1980;56(3):409-416.
Modern review:
- Berentsen S. How I treat cold agglutinin disease. Blood. 2021 Mar 11;137(10):1295-1303.