Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
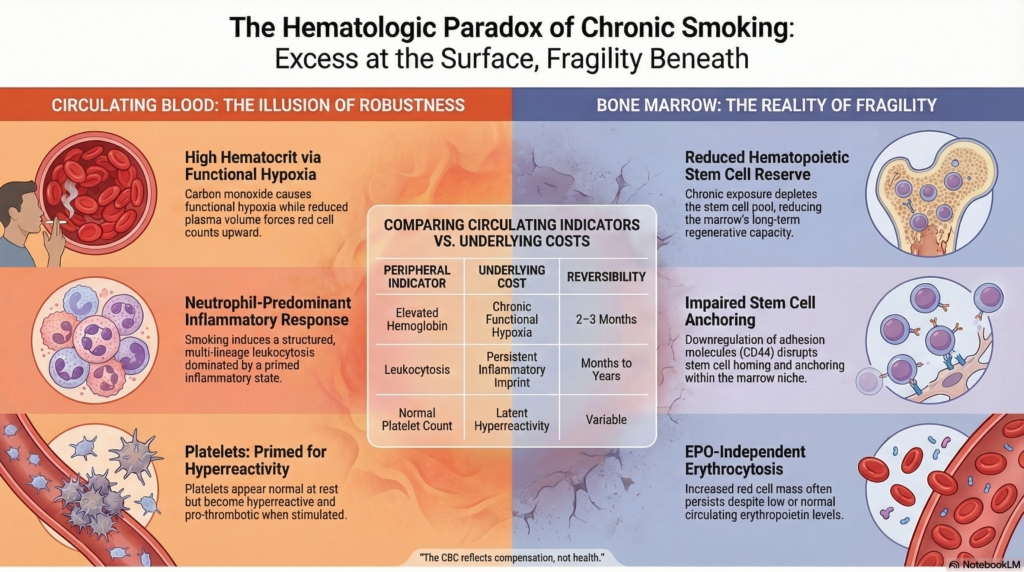
Smoking produces a paradox in blood.1
At the level of circulating cells, it creates excess: higher hemoglobin, higher white blood cell counts, more active platelets. The system appears robust. Yet at the level of its regulation and reserve, the same exposure imposes constraint. Hematopoiesis becomes stressed, inflammatory signaling persists, and the capacity to respond to future demands is altered.2
This tension, between apparent strength and underlying fragility, organizes the hematologic effects of smoking. What we measure in the CBC is not simply a set of abnormalities, but a record of chronic exposure, compensation, and cost.
Red blood cells: two mechanisms, one phenotype
Erythrocytosis is a common hematologic complication of smoking. It is often presented as a straightforward response to hypoxia. It is not. Two distinct mechanisms operate simultaneously, and the clinical picture reflects their combination.3
Carbon monoxide binds hemoglobin with high affinity, reducing oxygen content while also shifting the oxygen dissociation curve to the left, impairing oxygen release to tissues.4 The consequence is dual: less oxygen carried and less oxygen released. This functional hypoxia stimulates erythropoiesis and increases red cell mass.5
At the same time, smoking can reduce plasma volume, producing a relative polycythemia.6 These are not competing explanations but coexisting processes. In physiologic studies, many smokers exhibit both increased red cell volume and reduced plasma volume, while others show only one component.7
The erythropoietin response is not linear. Once red cell mass increases, circulating erythropoietin levels may be normal or low, reflecting feedback rather than absence of stimulus. In smokers, erythrocytosis is not reliably accompanied by elevated erythropoietin, a feature that distinguishes it from other hypoxic states.
This process is tightly linked to carbon monoxide exposure. Reduction of carbon monoxide, through cessation or substitution with low carbon monoxide delivery systems, leads to normalization of hematocrit, often within two to three months.8 This reversibility is both diagnostic and therapeutic.
Importantly, this is not a benign adaptation. Secondary erythrocytosis in smokers carries thromboembolic risk, in some series comparable to that of primary polycythemia.9 The elevated hematocrit is not simply a number; it is a risk state.
Selected evidence
Study design and population
Retrospective epidemiological cohort study of 2,940 apparently healthy adults (1,503 men and 1,437 non-pregnant women), across age groups 30 to 60 years, with assessment of smoking and alcohol consumption by questionnaire.
Key findings
| Domain | Finding |
|---|---|
| Hemoglobin | Higher in smokers vs nonsmokers (both sexes) |
| Dose response | Positive correlation with cigarettes/day (significant in women) |
| Sex effect | Greater increase in women than men |
| Alcohol | Higher Hb with heavy alcohol use (>14 drinks/week men, >7 women) |
| Combined exposure | Smoking and alcohol effects are additive and correlated |
| Iron status | No consistent relationship with ferritin |
Interpretation
Smoking is associated with modest but consistent increases in hemoglobin within the normal range, with evidence of dose dependence, particularly in women. Alcohol exerts a similar, smaller effect, and the two exposures frequently co-occur, making their individual contributions difficult to fully separate. The mechanism is consistent with carbon monoxide–mediated functional hypoxia stimulating erythropoiesis.
Conclusion
In this large population cohort, smoking is associated with small, exposure-dependent increases in hemoglobin within the normal range, more pronounced in women, though the magnitude of change is insufficient to justify separate clinical reference ranges.
Study design and population
Cross-sectional study of 227 patients with elevated hemoglobin (≥160 g/L), evaluating the association between cigarette and shisha smoking and severity of polycythemia.
Key findings
| Domain | Finding |
|---|---|
| Hemoglobin | Higher in smokers vs nonsmokers |
| Smoking type | Highest Hb in combined cigarette + shisha users |
| Dose / severity | Greater smoking associated with higher Hb categories |
| Polycythemia | 53% of cohort had Hb >172 g/L |
| Risk association | Modestly increased odds of more severe polycythemia (OR ≈1.11) |
Interpretation
In this selected population with elevated hemoglobin, smoking is associated with higher hemoglobin levels and greater severity of polycythemia, with the strongest effect observed in combined cigarette and shisha users. These findings are consistent with hypoxemia-driven erythropoiesis related to carbon monoxide exposure.
Conclusion
Among patients with elevated hemoglobin, smoking is associated with incrementally higher hemoglobin levels and modestly greater polycythemia severity, supporting a role for smoking as a contributor to secondary erythrocytosis.
Study design and population
Physiologic study of 22 smokers with elevated hematocrit and elevated carboxyhemoglobin, evaluating red cell volume, plasma volume, and oxygen transport parameters.
Key findings
| Domain | Finding |
|---|---|
| Carboxyhemoglobin | Markedly elevated (mean ~11.6%) |
| Oxygen transport | Left shift of oxygen dissociation curve (↓ P50 with increasing COHb) |
| Red cell volume | Increased in a substantial subset (e.g., 12/18 measured patients) |
| Plasma volume | Reduced in most measured patients (14/18) |
| Blood volume | Variable |
| Reversibility | Red cell volume decreased and plasma volume increased after smoking reduction/cessation |
Interpretation
Smoking-related polycythemia reflects two concurrent mechanisms: increased red cell volume and reduced plasma volume. Carbon monoxide exposure reduces effective oxygen-carrying capacity and shifts the hemoglobin–oxygen dissociation curve to the left, impairing oxygen delivery to tissues and stimulating erythropoiesis.
These findings demonstrate a dissociation between circulating measurements and underlying physiology, in which hematocrit reflects both production and volume changes.
Conclusion
This study demonstrates that smoking-associated polycythemia arises from coordinated changes in oxygen transport, red cell production, and plasma volume, all driven by carbon monoxide exposure, and that these abnormalities are at least partially reversible with cessation.
Study design and population
Large population-based cohort (n = 6,808) from the PREVEND study, assessing smoking exposure (self-report and urinary cotinine) in relation to hematocrit, hemoglobin, MCV, and erythropoietin (EPO) levels.
Key findings
| Domain | Finding |
|---|---|
| Hemoglobin / hematocrit | ↑ in current smokers vs nonsmokers |
| Erythrocytosis | more prevalent in smokers (≈4% vs 1%) |
| EPO levels | ↓ in smokers (inverse association) |
| Dose-response | higher cotinine → lower EPO |
| MCV | ↑ in smokers |
| Multivariable analysis | smoking independently ↑ Hb/Hct and ↓ EPO |
Interpretation
Smoking is associated with higher hemoglobin and hematocrit despite lower circulating EPO levels, contradicting the conventional model of hypoxia-driven, EPO-mediated erythrocytosis.
Conclusion
Smoking-related erythrocytosis appears to be EPO-independent or EPO-suppressed, suggesting alternative mechanisms such as altered erythroid sensitivity, circadian EPO dynamics, or direct effects on erythropoiesis
White blood cells: a patterned inflammatory signal
If the red cell compartment reflects hypoxia, the white cell compartment reflects inflammation.
Smoking-associated leukocytosis has a recognizable phenotype: a multi-lineage expansion with internal structure. Neutrophilia is nearly universal, but it rarely occurs alone. Lymphocytosis, monocytosis, and basophilia frequently co-occur, forming a consistent differential pattern rather than a nonspecific elevation.10
This pattern reflects distinct biological drivers. Neutrophils increase disproportionately, while lymphocytes and eosinophils may decline as a proportion of total leukocytes despite absolute increases.11 Different lineages respond to different exposure dimensions, including cumulative burden, current intensity, and time since cessation, producing a structured, not random, differential.12
Mechanistically, this is a chronic inflammatory leukocytosis. Smoking induces cytokine signaling, impairs alveolar macrophage clearance, and shortens neutrophil marrow transit time. The result is a persistent, exposure-linked inflammatory state.13
The temporal behavior is uneven. Leukocyte counts may decline after cessation, with measurable improvement over weeks and a median around two months in selected cohorts. But normalization is not assured.14 Elevated counts may persist for years after cessation, reflecting a durable inflammatory imprint.15
Clinically, tobacco-associated leukocytosis is a diagnosis of exclusion. Patients are often evaluated for clonal disease before attribution, including molecular testing and, in selected cases, marrow examination. Recognition of the pattern guides interpretation, but does not replace diagnostic rigor.
Selected evidence
Study design and population
Cross-sectional analysis of 6,138 adults aged 30 to 74 years from the First National Health and Nutrition Examination Survey (NHANES I), examining the relationship between smoking exposure and leukocyte differentials, adjusted for age, sex, race, and BMI.
Key findings
| Domain | Finding |
|---|---|
| Total WBC | ↑ with current smoking; also associated with pack-years and years since quitting |
| Neutrophils | ↑ (dominant contributor; disproportionately increased as % of WBC) |
| Lymphocytes | ↑ in absolute count; ↓ as % of total WBC |
| Monocytes | ↑ (associated primarily with current smoking intensity) |
| Eosinophils | ↑ (associated primarily with current smoking intensity) |
| Differential pattern | Neutrophil predominance with relative reduction in lymphocyte and eosinophil percentages |
| Dose-response | Nonlinear relationship with current cigarettes/day |
Interpretation
Smoking produces a structured leukocytosis, characterized by increases across multiple leukocyte lineages with a disproportionate rise in neutrophils. Absolute neutrophil and lymphocyte counts are associated with both current and cumulative exposure, whereas monocytes and eosinophils are linked primarily to current smoking intensity. At the same time, the relative composition shifts toward neutrophil predominance. These effects are driven mainly by current exposure, with smaller contributions from cumulative exposure and partial persistence after cessation.
Conclusion
Cigarette smoking is associated with a multi-lineage but non-uniform leukocytosis, dominated by neutrophilia and shaped primarily by current smoking intensity, with additional contributions from cumulative exposure and incomplete reversal after cessation.
Study design and population
Retrospective observational study of 40 patients referred to hematology for persistent leukocytosis, in whom tobacco use was determined to be the cause after exclusion of infection, inflammatory conditions, and clonal hematologic disorders.
Key findings
| Domain | Finding |
|---|---|
| Total WBC | Mild elevation (mean ~13.3 × 10⁹/L) |
| Neutrophils | ↑ in 98% (dominant feature) |
| Lymphocytes | ↑ in 53% |
| Monocytes | ↑ in 50% |
| Basophils | ↑ in 48% |
| Eosinophils | ↑ in 23% |
| Platelets | ↑ in 20% |
| Red cells | Erythrocytosis in 30% |
| Reversibility | WBC ↓ with smoking reduction/cessation (mean 13.2 → 11.1 × 10⁹/L) |
| Time course | Median 8 weeks to improvement (range 2–49 weeks) |
Interpretation
Smoking-associated leukocytosis presents as a mild elevation in white blood cell count with a structured, multi-lineage pattern, dominated by neutrophilia but frequently accompanied by lymphocytosis, monocytosis, and basophilia. In patients referred for evaluation of unexplained leukocytosis, tobacco use may account for the abnormality after exclusion of other causes.
Importantly, leukocytosis is reversible, with improvement typically observed within weeks of smoking reduction or cessation, supporting a direct and dynamic relationship between exposure and hematologic phenotype.
Conclusion
Tobacco use is a reversible cause of mild, structured leukocytosis, characterized by neutrophil predominance and multi-lineage involvement, and should be considered in the evaluation of asymptomatic leukocytosis before pursuing extensive diagnostic testing.
Study design and population
Cross-sectional study of 2,511 male workers aged 25 to 62 years, evaluating associations between smoking status, time since cessation, and hematologic parameters using multivariable analysis.
Key findings
| Domain | Finding |
|---|---|
| Total WBC | ↑ in current smokers (OR ≈12.1 for WBC ≥9,000/mm³) |
| Persistence | WBC remains elevated in ex-smokers 5–9.9 years after cessation (OR ≈3.8) |
| Hemoglobin | Modestly ↑ in current smokers (OR ≈1.6 for Hb ≥16 g/dL) |
| Dose-response | WBC and Hb ↑ with cigarettes/day (highest in ≥20/day group) |
| Alcohol | Lower odds of leukocytosis with higher intake (OR ≈0.4 for ≥14 units/week) |
| Covariates | Associations persist after adjustment for age, BMI, BP, activity |
Interpretation
Smoking is associated with marked leukocytosis and modest increases in hemoglobin, with clear dose-response effects. Importantly, leukocytosis may persist for at least 5–9 years after smoking cessation in some individuals, indicating that the inflammatory effects of smoking are not immediately reversible. In contrast, the effect on hemoglobin appears limited to current smoking, suggesting different temporal dynamics across cell lines.
Conclusion
Cigarette smoking produces leukocytosis that may persist for years after cessation, alongside more transient effects on hemoglobin, supporting a model in which smoking induces a durable inflammatory imprint on the hematologic system.
Platelets and coagulation: function over number
Smoking shifts hemostasis toward thrombosis, but not through platelet count.
Platelet numbers are often normal. What changes is behavior. Platelets exist in a primed state, appearing quiescent at rest but demonstrating increased aggregation when challenged. This latent hyperreactivity is accompanied by increased thromboxane generation, linking platelet function directly to exposure intensity.16
At the same time, the surrounding hemostatic environment is altered. Fibrinogen rises as part of an IL-6–driven acute-phase response, and fibrinolysis is impaired, in part through increased expression of plasminogen activator inhibitor-1 (PAI-1). Endothelial dysfunction further amplifies these effects, creating a prothrombotic substrate.
These processes converge. Red cell excess increases viscosity, platelet reactivity promotes clot formation, and endothelial injury provides the surface on which thrombosis occurs. Dyslipidemia contributes additional vascular injury.
The key point is that counts do not reflect function. A normal platelet count in a smoker does not imply normal hemostasis. The abnormality is not constant activation, but a system that is more likely to clot when stressed.
Selected evidence
Study design and population
Cross-sectional, multicenter observational study (Total Exposure Study) including 3,585 adult smokers and 1,077 nonsmokers, examining relationships between biomarkers of smoke exposure and biomarkers of inflammation, oxidative stress, and platelet activation.
Key findings
| Domain | Finding |
|---|---|
| Inflammation | ↑ WBC, ↑ hs-CRP, ↑ fibrinogen, ↑ vWF in smokers vs nonsmokers |
| Dose-response | WBC, hs-CRP, fibrinogen ↑ with cigarettes/day and exposure biomarkers |
| Oxidative stress | ↑ 8-epiPGF2α in smokers; ↑ with exposure intensity |
| Platelet activation | ↑ 11-dehydro-TxB2 in smokers; ↑ with cigarettes/day and exposure biomarkers |
| Exposure linkage | Biomarkers of harm correlate with nicotine metabolites, NNAL, COHb, and other exposure markers |
| Magnitude | Effects modest; models explain ≤22% of variability |
| Confounders | BMI is a dominant driver across multiple biomarkers |
Interpretation
Smoking is associated with a coordinated biological response across inflammation, oxidative stress, and platelet activation, rather than isolated abnormalities. Platelet activation, reflected by increased thromboxane production, rises in parallel with exposure intensity and inflammatory signaling. These relationships track with internal biomarkers of smoke exposure, supporting a direct link between exposure burden and downstream vascular and hematologic effects.
At the same time, the modest explanatory power of these models highlights the multifactorial nature of these pathways, with factors such as BMI contributing substantially to variability.
Conclusion
Cigarette smoking is associated with integrated activation of inflammatory, oxidative, and platelet pathways, with dose-dependent relationships to exposure, supporting a model in which smoking promotes a prothrombotic and proinflammatory state through multiple converging mechanisms.
Study design and population
Cross-sectional physiologic study of 20 healthy chronic smokers and 23 nonsmokers, evaluating platelet adhesion, activation, aggregation, and release markers using electron microscopy and in vitro activation assays.
Key findings
| Domain | Finding |
|---|---|
| Platelet count | No significant difference |
| Circulating activation | No difference in platelet aggregate ratio, platelet factor 4, or β-thromboglobulin |
| In vitro aggregation | ↑ platelet aggregates in smokers (80 vs 43 per 100 platelets, p<0.01) |
| Exposure effect | Greater aggregation with longer smoking duration (≥20 years) |
| Age effect | Greater aggregation in smokers ≥50 years |
| Dose (packs/day) | No clear difference by daily quantity |
Interpretation
Chronic smoking is associated with enhanced platelet reactivity in response to an activating stimulus, despite normal platelet counts and no detectable increase in circulating activation markers. Platelets appear functionally “primed,” exhibiting increased aggregation only when challenged in vitro. This creates a dissociation between circulating measurements and functional capacity.
Conclusion
Smoking is associated with platelet priming rather than overt activation, such that platelets behave normally at rest but demonstrate increased aggregation when stimulated, providing a mechanistic basis for increased thrombotic risk despite normal platelet counts.
Bone marrow and hematopoiesis: stress beneath stability
The most revealing effects of smoking may lie upstream, in the marrow itself.
Experimental and translational models show that hematopoietic stem and progenitor cell populations can be reduced even while peripheral blood counts remain unchanged. Importantly, this effect is not limited to whole cigarette smoke. Studies of inhaled nicotine-containing aerosol demonstrate that exposure alone is sufficient to perturb hematopoietic biology, establishing the plausibility of marrow effects independent of combustion products.
This creates a dissociation between compartments. The circulating blood may appear stable, while the production system is already under strain.
That strain operates along several dimensions. First, the stem cell pool itself is reduced, with fewer hematopoietic stem and progenitor cells and impaired engraftment capacity. Second, the marrow microenvironment is altered. Mesenchymal stromal cells, which form the structural and signaling backbone of the stem cell niche, are decreased in number and function, and exhibit dysregulated signaling that can paradoxically promote progenitor proliferation despite reduced reserve. Third, the spatial organization of hematopoiesis is disrupted. Adhesion molecules such as CD44 are downregulated on stromal and endothelial cells, impairing stem cell homing and anchoring within the niche. The problem is not only that there are fewer stem cells, but that those that remain cannot properly engage the environment that sustains them.
Under baseline conditions, functional output is often preserved. But under stress, particularly inflammatory challenge, these latent changes emerge. In experimental models, hematopoietic stem cells from exposed animals show myeloid-skewed output after stimulation, with increased monocyte production following transplantation. The skewing is not a resting-state phenomenon; it is a stress-revealed phenotype. That latency is the finding.
At the molecular level, smoking is associated with shortened leukocyte telomere length, reflecting accelerated cellular aging. This effect is dose-dependent and only partially reversible over time, consistent with cumulative injury to the hematopoietic system.
Taken together, these findings suggest that smoking imposes chronic stress on hematopoiesis, reducing reserve, altering response to challenge, and disrupting the architecture that sustains blood production. The marrow is not simply suppressed or stimulated. It is dysregulated. This framework also provides a biologic link to epidemiologic associations between smoking and myeloproliferative neoplasms, in which clonal expansion may be favored in a stressed and disordered marrow environment.
Selected evidence
Study design and population
Experimental murine study of chronic cigarette smoke exposure (6 hours/day, 5 days/week for 9 months), examining effects on hematopoietic stem and progenitor cells (HSPCs) and the bone marrow niche, including mesenchymal stromal cells (MSCs) and engraftment capacity.
Key findings
| Domain | Finding |
|---|---|
| HSPCs | ↓ Lin−c-kit+Sca-1+ stem/progenitor cells |
| LT-HSCs | ↓ trend in long-term repopulating HSCs |
| MSCs (niche cells) | ↓ number and ↓ colony-forming capacity |
| Engraftment | ↓ HSPC engraftment in smoke-exposed marrow |
| Niche signaling | ↓ Angpt1, BMP4, CXCL12, Dkk2; ↑ Jag1, PDGFα |
| Co-culture behavior | Smoke-exposed MSCs → ↑ HSPC proliferation |
| System behavior | Reduced stem cell pool despite proliferative signaling |
Interpretation
Chronic cigarette smoke exposure reduces hematopoietic stem cell number and impairs the bone marrow niche, particularly through depletion and functional alteration of mesenchymal stromal cells. Despite this reduction in stem cell pool and engraftment capacity, smoke-exposed niches exhibit pro-proliferative signaling that drives expansion of progenitor cells, suggesting dysregulated rather than suppressed hematopoiesis.
Conclusion
Smoking is associated with quantitative loss and qualitative dysfunction of the hematopoietic stem cell compartment, characterized by reduced reserve, impaired niche support, and paradoxical proliferative signaling, providing a mechanistic basis for simultaneous peripheral cytosis and reduced marrow resilience.
Study design and model
In vivo cigarette smoke exposure and in vitro nicotine exposure using long-term bone marrow cultures (LTBMCs), assessing effects on hematopoiesis, stromal cells, adhesion molecules, and stem cell homing.
Key findings
| Domain | Finding |
|---|---|
| Hematopoiesis | ↓ myeloid progenitors in bone marrow (≈2-fold reduction) |
| Stem cell reserve | ↓ long-term culture-initiating cells (LTC-ICs) |
| Niche formation | impaired “cobblestone areas” (functional niches) |
| Stromal biology | nicotine disrupts stromal support, not progenitor proliferation |
| Adhesion molecules | ↓ CD44 expression on stromal and endothelial cells |
| Homing | ↓ HSPC homing to bone marrow (~6-fold reduction in CFU-S) |
| Platelets | delayed platelet recovery after transplant |
| Mechanism | impaired stem cell–stroma adhesion and trafficking |
Interpretation
Nicotine and cigarette smoke impair hematopoiesis primarily by disrupting the bone marrow microenvironment, rather than directly inhibiting progenitor proliferation. This occurs through downregulation of key adhesion molecules (especially CD44) and impairment of stromal cell function, leading to defective niche formation and reduced stem cell homing.
Conclusion
Smoking interferes with hematopoiesis at the level of stem cell anchoring and trafficking, reducing effective stem cell seeding and niche interactions, thereby limiting marrow regenerative capacity despite preserved proliferative potential of committed progenitors.
Exposure, duration, and heterogeneity
The hematologic effects of smoking vary with intensity, duration, and biological context.
Leukocytosis may not emerge until several years of exposure.17 Red cell mass increases more clearly with higher intensity smoking. Some populations show substantial elevations in hemoglobin and hematocrit, while others show more modest changes,18 reflecting differences in exposure and baseline physiology.
Sex differences are clinically relevant. In several cohorts, erythrocytosis and leukocytosis are more pronounced in men, affecting how abnormalities are recognized relative to sex-specific reference ranges.19
The CBC reflects the cumulative interaction of these variables rather than a single uniform response.20
Clinical perspective
The hematologic profile of smoking is recognizable but must be interpreted in context.
Elevated hemoglobin often reflects secondary erythrocytosis but requires exclusion of primary disease.21 Leukocytosis may reflect chronic inflammation but must be distinguished from clonal processes.22 Platelet counts may be normal despite increased thrombotic risk.
At the same time, these findings create an opportunity. Attribution of abnormalities to smoking can serve as a powerful moment for intervention, with measurable rates of smoking reduction when counseling is delivered in a hematologic context.
Closing perspective
Smoking does not produce a single hematologic disorder. It reorganizes the system across levels.
At the surface, circulating cells increase. Hemoglobin rises, leukocytes expand, and platelets become more reactive. The blood appears active, even overactive. Yet beneath this apparent strength, the mechanisms that sustain it—hematopoiesis, immune regulation, and vascular integrity—are altered. The marrow contains fewer stem cells, the niche that supports them is disrupted, and the signals that govern their behavior are distorted. Platelets appear normal at rest but respond excessively when challenged. Leukocytosis reflects not only inflammation, but a structured and persistent shift in immune composition. Even erythropoiesis becomes uncoupled from its canonical regulator.
What we see in the CBC is therefore not simply activation, but compensation within a system that has lost flexibility.
The result is a different kind of stability, one that holds under ordinary conditions and reveals its limits under stress.
In this setting, the CBC is not just a measurement. It is a record of exposure—and of the cost of adaptation.
Selected Evidence
Study design and population
Cross-sectional study of 156 healthy adults (56 smokers, 100 nonsmokers). Smokers consumed 10–20 cigarettes per day for at least 3 years.
Key findings
| Domain | Finding |
|---|---|
| Red cells | Hemoglobin ↑, hematocrit ↑, RBC ↑ (particularly in males) |
| Red cell indices | MCV ↑, MCH ↑ |
| White cells | WBC ↑ |
| Platelets | No significant difference |
| Other indices | MCHC, RDW largely unchanged |
Interpretation
Smoking is associated with higher hemoglobin, hematocrit (particularly in men), macrocytosis, and leukocytosis, consistent with combined hypoxia-driven erythropoiesis and chronic inflammatory activation.
Conclusion
In this cohort, sustained cigarette smoking is associated with higher hemoglobin, hematocrit (in men), leukocytosis, and increased red cell indices, supporting a model of exposure-driven hematologic activation with potential implications for cardiovascular and thrombotic risk.
Study design and population
Cross-sectional study of 80 healthy male subjects (40 smokers, 40 nonsmokers), aged 25 to 40 years. Smoking intensity was quantified using a smoking index and stratified into mild, moderate, and heavy exposure.
Key findings
| Domain | Finding |
|---|---|
| Red cells | Hemoglobin ↑, hematocrit ↑ |
| Red cell count | RBC ↑ with increasing smoking intensity |
| White cells | WBC ↑ |
| Differential | Neutrophils ↑ with higher intensity |
| Platelets | No significant difference |
| Lipids (contextual) | Atherogenic profile worsens with intensity |
Interpretation
Smoking is associated with increased hemoglobin, hematocrit, and leukocytosis, with consistent intensity-dependent effects. Heavier smoking is linked to progressive increases in red cell count and total leukocyte count, with a specific rise in neutrophils, supporting a graded relationship between exposure and both hypoxic and inflammatory responses.
Conclusion
This study demonstrates that the hematologic effects of smoking are dose-dependent, with heavier exposure producing more pronounced elevations in red cell mass surrogates and inflammatory leukocytosis, reinforcing the link between smoking intensity and cardiovascular risk.
Study design and population
Cross-sectional study of 171 healthy young men aged 20 to 30 years, comparing smokers (n=101) and nonsmokers (n=70), with subgroup analysis based on duration of smoking.
Key findings
| Domain | Finding |
|---|---|
| Red cells | Hemoglobin and hematocrit: no significant difference |
| Red cell indices | MCV ↑ in smokers |
| White cells | No overall difference; WBC ↑ with ≥5 years of smoking |
| Platelets | No significant difference |
| Iron / nutritional | Ferritin, iron, B12, folate unchanged |
Interpretation
In this young, otherwise healthy population, smoking is associated primarily with macrocytosis and duration-dependent leukocytosis, without consistent changes in hemoglobin, hematocrit, or platelet count. The effect on white blood cells emerges with longer exposure, supporting a time-dependent inflammatory response rather than an immediate change.
Conclusion
In this cohort, cigarette smoking is associated with subtle, exposure-dependent hematologic changes, characterized by increased MCV and leukocytosis with longer duration of smoking, while other hematologic parameters remain largely unchanged.