Why size, force, and cleavage determine VWF behavior
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
Why this spoke matters
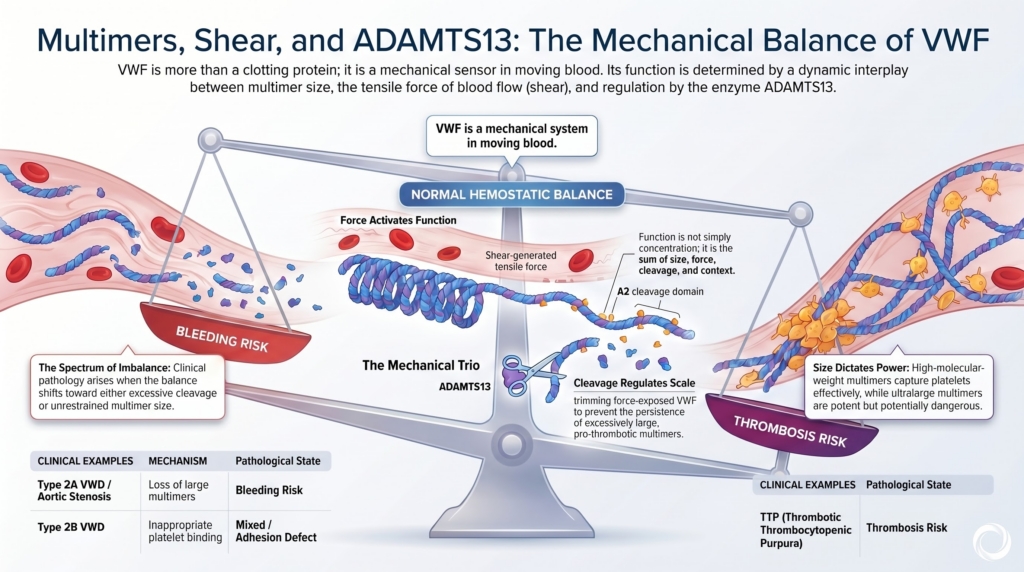
VWF is not simply present or absent. It has size, shape, tension, and mechanical behavior. It changes under force. It binds platelets when stretched. It is trimmed when unfolded.
This is why multimers, shear, and ADAMTS13 matter.
They explain:
- why the largest VWF forms are so hemostatically powerful
- why type 2A VWD causes bleeding
- why type 2B can bleed despite increased platelet binding
- why aortic stenosis and LVADs can cause acquired VWF abnormality
- why TTP sits at the thrombotic edge of the same VWF–ADAMTS13 axis
The central question is:
How does VWF remain adhesive enough to stop bleeding, but regulated enough to avoid thrombosis?
The answer is size, force, and cleavage.
The importance of size
VWF circulates as multimers. These range from smaller forms to very large high-molecular-weight multimers and ultralarge forms released from endothelial storage.
The largest multimers are not merely bigger. They are functionally different. They contain more binding sites, respond more dramatically to flow, form longer adhesive strings, and are especially effective at recruiting platelets under high shear.1
This is why multimer size is clinically important. A patient may have measurable VWF antigen but still lack the largest, most hemostatically useful forms.
The laboratory may show:
VWF present.
The vessel wall may experience:
VWF inadequate.
That is the multimer lesson.
Multimer size is a physiologic property
It is tempting to treat multimer analysis as a specialized laboratory add-on. But multimer size is not just a laboratory pattern. It is a physiologic property.
VWF must work in flowing blood. In low-flow settings, adhesive interactions can occur through slower contact. In high-flow settings, platelets need to be captured rapidly. Large VWF multimers are built for that task. They create a multivalent adhesive platform that can bind exposed matrix and tether platelets moving at speed.
This is why loss of high-molecular-weight multimers can produce bleeding that seems disproportionate to the antigen level.2
The question is not only:
How much VWF is there?
It is:
What size is it, and how does it behave under force?
VWF as a mechanosensor
VWF is unusual among hemostatic proteins because it is regulated not only by concentration or chemical activation, but by mechanical force.
It is a protein designed to feel the bloodstream.
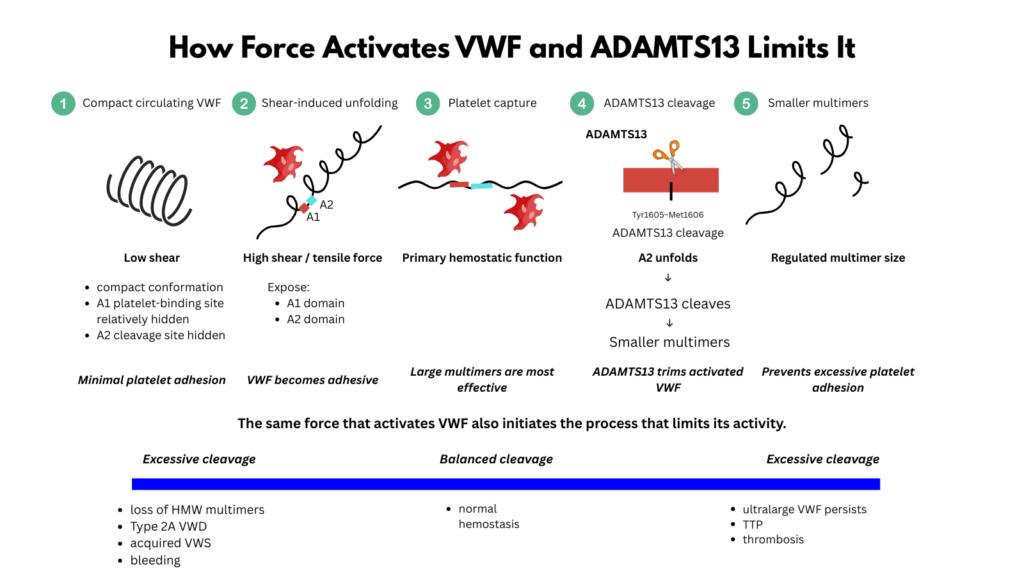
In relatively low shear, VWF circulates in a compact or loosely coiled state. Its platelet-binding function is restrained. This is essential. If VWF were fully adhesive everywhere, normal circulation would become dangerous.
At sites of vascular injury, in the microcirculation, or across pathological high-shear lesions, VWF elongates. Shear-generated tensile force changes its function. It exposes or enhances platelet-binding interactions, especially through the A1 domain and platelet glycoprotein Ibα. It also unfolds the A2 domain, which contains the Tyr1605-Met1606 bond cleaved by ADAMTS13.3
This is the paradox and brilliance of the system:
the force that activates VWF also makes it cleavable.
Shear turns VWF on and turns it down
Shear has two opposing effects on VWF.
It activates VWF by stretching the molecule and promoting platelet capture. But it also permits ADAMTS13 to trim VWF by unfolding the A2 domain and exposing the cleavage site.
This is not a contradiction. It is homeostasis.
At a site of injury, VWF must become adhesive quickly enough to catch platelets. But once ultralarge VWF enters the circulation, its adhesive potential must be limited. ADAMTS13 performs that editing function.
ADAMTS13 is active in plasma. The key regulated event is not classic protease activation, but substrate exposure: VWF becomes cleavable when force exposes A2.4
Too little VWF function causes bleeding. Unregulated ultralarge VWF is thrombogenic.
VWF lives between those dangers.
ADAMTS13 as an editor
ADAMTS13 is often described as a VWF-cleaving protease. That is true, but incomplete.
ADAMTS13 is better understood as a size editor.
It does not erase VWF. It trims it.
ADAMTS13 edits VWF at the endothelial surface, where newly released ultralarge VWF strings are processed, and in the circulation, where force-dependent unfolding targets the largest multimers. This editing converts newly released, highly adhesive VWF into a circulating distribution of multimers compatible with normal hemostasis.5
The word “cleavage” can sound destructive.
In VWF biology, cleavage is regulatory.
The A2 domain: the force-sensitive hinge
The A2 domain is central to this system. It contains the Tyr1605-Met1606 bond cleaved by ADAMTS13. In the folded state, this site is relatively protected. Under tensile force, A2 unfolds, making the cleavage site accessible.
Unlike adjacent A domains, A2 lacks a stabilizing disulfide bond, making it especially susceptible to force-induced unfolding.6
This is why ADAMTS13 cleavage is force-dependent. The protease does not simply attack all circulating VWF indiscriminately. It preferentially cleaves VWF that has been mechanically stretched. Longer multimers experience greater tensile forces and are therefore more likely to unfold and become substrates for ADAMTS13.
This creates an elegant feedback loop:
- the largest multimers are the most adhesive
- the largest multimers experience the most force
- the largest multimers are most likely to expose A2
- exposed A2 permits ADAMTS13 cleavage
- cleavage reduces adhesive potential
The molecules most capable of causing thrombosis are also the molecules most likely to be trimmed.
Why ultralarge VWF does not normally circulate
Endothelial cells store VWF in Weibel-Palade bodies. When released, VWF can emerge as ultralarge multimers or long strings. These forms are extremely adhesive.
That is useful at injury sites.
It is dangerous in the wrong place.
Ultralarge VWF is therefore normally processed rapidly. ADAMTS13 prevents it from persisting in a highly adhesive form in the circulation. Without ADAMTS13, ultralarge VWF can accumulate and bind platelets under shear, leading to the platelet-rich microvascular thrombosis of thrombotic thrombocytopenic purpura.7
TTP is not simply “too much clotting.”
It is failure to restrain a mechanically activated adhesive protein.
Type 2A: when large multimers are lost
Type 2A VWD is the inherited VWD subtype most directly tied to loss of high-molecular-weight multimers.
The result is impaired platelet-dependent adhesion under shear.
But type 2A is not one mechanism. Large multimers may be missing because:
- they are not assembled properly
- they are poorly secreted
- they are retained intracellularly
- they are excessively susceptible to ADAMTS13 cleavage after secretion
- their storage and regulated release are abnormal
Different molecular routes converge on the same structural endpoint:
loss of large multimers and impaired hemostatic function.8
Some type 2A mutations destabilize the A2 domain. The force required to expose the ADAMTS13 cleavage site falls. ADAMTS13 gains easier access, high-molecular-weight multimers are lost, and hemostatic function falls.9
Type 2A is best understood as a structural endpoint, not a single molecular story.
Type 2B: too much binding can produce bleeding
Type 2B VWD seems paradoxical. The defect increases VWF affinity for platelet GPIbα. One might expect increased binding to produce thrombosis.
Instead, patients bleed.
Why? Because excessive VWF-platelet interaction removes the most hemostatically effective VWF multimers from circulation. VWF-platelet complexes may be cleared or sequestered, contributing to both thrombocytopenia and loss of high-molecular-weight multimers. The result is reduced functional VWF and sometimes a reduced platelet count.10
The mechanism is hyperadhesion.
The phenotype is bleeding.
Type 2B teaches that hemostasis requires regulated adhesion. Too little binding causes bleeding. Too much inappropriate binding can also cause bleeding.
Type 2M and type 2N: defining the boundary of the axis
The multimer-shear-ADAMTS13 axis explains much of VWF biology, but not every VWD subtype sits on it in the same way.
Type 2M usually preserves multimer size but impairs VWF interaction with platelet GPIb or collagen. The problem is not absence of large multimers, but abnormal behavior of otherwise present multimers.
Type 2N is more orthogonal to the shear axis. Platelet adhesion and multimer pattern may be relatively preserved, but VWF binding to FVIII is impaired, producing a hemophilia A-like phenotype with reduced FVIII.11
The axis is powerful.
It is not the whole map.
Acquired high-shear VWF loss
Inherited VWD is not the only setting where high-molecular-weight multimers disappear.
High-shear cardiovascular conditions can mechanically alter VWF and increase susceptibility to proteolysis. Aortic stenosis is the classic example. As blood accelerates through a narrowed valve, shear stress increases. VWF unfolds more readily. ADAMTS13 cleavage increases. High-molecular-weight multimers are depleted. A type 2A-like acquired VWF abnormality can result.
This is acquired von Willebrand syndrome, not inherited VWD.
The key study by Vincentelli and colleagues showed that hemostatic abnormalities were frequent in severe aortic stenosis, correlated with valve severity, and improved after valve replacement when the mechanical shear problem was relieved.12
This is structure-function reasoning at the bedside.
Change the flow, and the VWF changes.
Heyde syndrome: the clinical bridge
Heyde syndrome links aortic stenosis, acquired VWF abnormality, and gastrointestinal bleeding from angiodysplasia.13
The bleeding is not explained by low platelet count or ordinary coagulation factor deficiency. It reflects loss of the largest VWF multimers in a high-shear environment, often compounded by fragile mucosal vascular lesions.
The concept matters because replacement alone may be temporary. If the shear lesion persists, newly supplied or released VWF may be cleaved again. Treating the valve can treat the VWF problem by removing the mechanical driver.
Sometimes the hemostatic treatment is anatomical.
LVAD-associated acquired VWF loss
Left ventricular assist devices create another high-shear environment. Blood passing through the device is exposed to nonphysiologic mechanical forces. VWF unfolds, high-molecular-weight multimers are lost, and an acquired VWF defect can develop.
This helps explain why patients with LVADs may develop mucosal and gastrointestinal bleeding, often with angiodysplasia-like lesions.14 The device supports circulation but may disrupt VWF structure.
The same axis appears again:
flow, unfolding, cleavage, loss of large multimers, bleeding.
TTP: the thrombotic edge
TTP sits at the thrombotic edge of the VWF-ADAMTS13 system.
In type 2A VWD and high-shear acquired VWF loss, the problem is loss of large multimers or excessive cleavage.
In TTP, the problem is severe ADAMTS13 deficiency.
Uncleaved ultralarge VWF persists. Platelets bind. Microvascular thrombi form. Thrombocytopenia, hemolysis, and organ injury follow.
Too much effective cleavage or loss of large multimers can produce bleeding.
Too little VWF trimming can produce platelet-rich microvascular thrombosis.
The same molecule sits between them.
Why laboratory tests are only approximations
VWF testing tries to translate force-dependent biology into static laboratory values.
That is difficult.
VWF antigen measures quantity. Platelet-dependent activity assays estimate platelet-binding function. Collagen-binding activity may be sensitive to high-molecular-weight multimer loss. Multimer analysis visualizes size distribution. FVIII level reflects VWF’s carrier function. RIPA and genetic testing can help classify type 2B. FVIII-binding assays help identify type 2N.15
Most assays are performed under static or simplified flow conditions. They cannot fully reproduce vessel injury, endothelial secretion, collagen exposure, platelet tethering, shear gradients, elongational flow, thrombin generation, fibrin deposition, and local ADAMTS13 regulation.
VWF is also context-sensitive. Levels may change with inflammation, stress, exercise, pregnancy, age, blood group, and preanalytic handling. Diagnostic guidance therefore emphasizes testing under baseline conditions, careful sample handling, repeat testing when appropriate, and relational interpretation across antigen, activity, FVIII, ratios, collagen binding, and multimer pattern.16
That does not make the assays unhelpful.
It makes them partial.
The clinician must interpret assays as approximations of VWF behavior, not direct replicas of in vivo hemostasis.
How guidelines operationalize the axis
Current diagnostic guidance implicitly uses the multimer-shear-ADAMTS13 axis.
VWF antigen asks how much protein is present. Platelet-dependent activity asks whether VWF can bind platelets. Collagen binding and multimer analysis help infer whether high-molecular-weight multimers are present. Activity-to-antigen ratios help distinguish quantitative deficiency from qualitative dysfunction. FVIII binding identifies type 2N, which sits outside the usual platelet-adhesion axis. Genetic testing helps resolve selected subtype questions, especially type 2B and 2N.17
The tests are not random.
They are attempts to infer size, adhesion, carrier function, and cleavage behavior.
Active VWF as a clinical window
Most circulating VWF is not fully adhesive at baseline. But in some settings, a fraction of VWF circulates in a more active conformation, more capable of binding platelet GPIb.
Assays of “active VWF” aim to detect this conformation. They provide a potential clinical window into force-dependent or inflammation-associated VWF activation and may help explain thrombotic risk in selected disorders.18
This concept reinforces the larger point:
VWF function depends not only on how much protein is present, but on what conformation it occupies.
Why this matters for treatment
Treatment can be understood through multimer-force biology.
Desmopressin releases endogenous VWF, including large multimers, if the patient has releasable stores. VWF concentrates replace missing or dysfunctional VWF, but products differ in multimer distribution and FVIII content. VWF-only products and recombinant VWF raise kinetic and structural questions about FVIII stabilization and multimer composition.
In high-shear acquired VWF loss, replacing VWF may help transiently, but the mechanical shear problem may continue to destroy the largest multimers. In TTP, the solution is not more VWF but restoration or replacement of ADAMTS13 activity and control of inhibitors.19
Mechanism determines therapy.
The molecule’s size and force behavior are not academic details.
They are treatment logic.
A single axis, many diseases
The VWF-ADAMTS13 shear axis helps connect multiple clinical entities:
- type 2A VWD
- type 2B VWD
- acquired VWS in aortic stenosis
- acquired VWS with LVADs
- GI bleeding from angiodysplasia in VWF disorders
- TTP
- broader thrombo-inflammatory states where VWF and ADAMTS13 balance may be altered
These are not the same disease. But they share a structural language:
- VWF size
- mechanical force
- platelet adhesion
- proteolytic trimming
- hemostatic balance
Understanding this axis makes the field feel less fragmented.
Clinical synthesis
VWF works because it is large, multimeric, adhesive, and force-responsive. The largest multimers are the most hemostatically effective, especially under shear. Shear stretches VWF and enables platelet capture, but also unfolds A2 for ADAMTS13 cleavage. ADAMTS13 trims VWF to control multimer size and prevent excessive platelet adhesion.
Bleeding occurs when effective VWF is too little, too small, too dysfunctional, too easily cleared, or too easily cleaved. Thrombosis occurs when ultralarge VWF adhesive potential is insufficiently restrained.
Type 2A VWD, type 2B VWD, acquired VWF loss in aortic stenosis or LVADs, and TTP all make sense along this same axis.
The central lesson is simple:
VWF is not only a clotting protein. It is a mechanical system in moving blood.
Evidence anchor: why size, force, and cleavage matter
Summary derived from structural biology, flow studies, single-molecule force experiments, multimer analyses, clinical VWD subtype studies, acquired VWS literature, and diagnostic guidance. The evidence consistently shows that VWF function depends on multimer size, force-dependent unfolding, and ADAMTS13-mediated regulation.
| Evidence stream | What it shows | Why it matters | Main limitation |
|---|---|---|---|
| Multimer biology | Larger VWF multimers have greater platelet- and collagen-binding capacity and are especially important under shear.20 | VWF quantity alone cannot define hemostatic function. Loss of high-molecular-weight multimers can cause bleeding despite measurable antigen. | Multimer analysis is technically demanding and not always rapidly available. |
| Shear-induced unfolding | Hydrodynamic force changes VWF conformation, enhancing platelet-binding capacity and making A2 accessible for cleavage.21 | VWF is a mechanosensitive protein, not a static adhesive molecule. Flow is part of its regulation. | Experimental flow systems simplify the complexity of vascular injury. |
| A2 domain force sensitivity | Single-molecule and molecular dynamics studies show that tensile force unfolds A2 and exposes the ADAMTS13 cleavage site.22 | ADAMTS13 cleavage is substrate-gated. It is targeted to mechanically stretched VWF. | Molecular systems isolate mechanisms that must be integrated with whole-blood biology. |
| ADAMTS13 regulation | ADAMTS13 trims ultralarge VWF and restrains excessive platelet adhesion; severe deficiency causes TTP.23 | The same axis explains bleeding from loss of large multimers and thrombosis from failure of trimming. | ADAMTS13 activity is only one determinant of thrombosis risk in inflammatory illness. |
| Type 2A mechanisms | Type 2A can arise from impaired multimer assembly, secretion, storage, or increased ADAMTS13 susceptibility.24 | Type 2A is a convergent structural phenotype: loss of high-molecular-weight multimers. | Genotype does not always predict bleeding severity for an individual patient. |
| Acquired high-shear VWF loss | Aortic stenosis and LVADs can produce acquired loss of high-molecular-weight multimers through shear-enhanced unfolding and proteolysis.25 | Flow can create a VWF disorder. In some patients, treating the mechanical lesion treats the hemostatic defect. | Bleeding often reflects multiple contributors, including angiodysplasia, anticoagulation, age, and comorbidity. |
| Diagnostic guidance | Guidelines emphasize VWF antigen, platelet-dependent activity, FVIII, ratios, collagen binding, multimer analysis, and subtype-specific testing when needed.26 | Assays interrogate different parts of VWF biology and must be interpreted relationally. | Laboratory tests are approximations of force-dependent biology in vivo. |
Interpretive note: The evidence supports a unified model: VWF function is created by size, activated by force, and restrained by ADAMTS13. Bleeding and thrombosis emerge when this balance is displaced in different directions.
Clinical interpretation guide: reading VWF through the multimer-shear-ADAMTS13 axis
Think beyond antigen
A normal or near-normal VWF antigen does not guarantee normal VWF function. Ask whether the largest multimers are present and whether activity is appropriate for antigen.
Read ratios as structure-function clues
Low platelet-dependent activity relative to antigen suggests qualitative dysfunction. Low collagen binding relative to antigen may suggest loss of high-molecular-weight multimers or impaired collagen binding.
Use multimer analysis to localize the problem
Loss of high-molecular-weight multimers points toward type 2A, type 2B, or acquired VWS. Normal multimers with low platelet-dependent activity suggests type 2M. Normal multimers with low FVIII out of proportion to VWF suggests type 2N.
Different multimer patterns may also help distinguish inherited and acquired processes, especially when interpreted with clinical history, platelet count, collagen binding, RIPA or genetic testing, and the activity-to-antigen ratio.
Remember high-shear acquired VWS
Consider acquired VWS when bleeding occurs with:
- severe aortic stenosis
- LVAD support
- other high-shear cardiac lesions
- unexplained GI bleeding or angiodysplasia
- new bleeding without a lifelong personal or family history
Do not confuse replacement with correction of force
VWF replacement may temporarily improve hemostasis in high-shear acquired VWS, but the underlying shear lesion may continue to remove high-molecular-weight multimers.
Use treatment logic
- Desmopressin depends on releasable endogenous VWF and a useful response.
- VWF concentrate replaces missing or dysfunctional VWF.
- Antifibrinolytics protect mucosal hemostasis.
- High-shear acquired VWS may require correction of the mechanical lesion.
- TTP requires restoration of ADAMTS13 activity and control of inhibitors.
Practical takeaway: In VWF biology, function is not simply concentration. It is concentration plus size, size plus force, force plus cleavage, and cleavage plus clinical context.
Reflect & Apply Case
A 78-year-old man develops recurrent melena from small-bowel angiodysplasia.
He has severe aortic stenosis.
Laboratory testing shows normal VWF antigen, reduced VWF collagen-binding activity, and loss of high-molecular-weight VWF multimers.
Questions for reflection:
- Why does a normal VWF antigen not exclude a clinically important VWF problem?
- What does loss of high-molecular-weight multimers imply functionally?
- How can aortic stenosis produce an acquired type 2A-like pattern?
- Why might replacing VWF provide only temporary benefit if the valve lesion persists?
- How does this case connect VWF structure, flow, ADAMTS13, and GI bleeding?
Key synthesis: High shear across the stenotic valve unfolds VWF, increases ADAMTS13 access to A2, depletes high-molecular-weight multimers, impairs platelet adhesion under shear, and contributes to bleeding from angiodysplasia.
This case illustrates the central lesson:
in VWF biology, flow can create the disease.
To treat the bleeding, sometimes the clinician must treat the force.
Test your thinking
A short, judgment-focused quiz on long-term management & monitoring in cold agglutinin disease.