“In regard to the functional activity of the spleen it will probably become more and more convenient to speak of ” hypersplenism” and- ” hyposplenism “- by analogy with the terms “hyperthyroidism” and ” hypotlhyroidism,” “hyperpituitarism ” and ” hypopituitarism.” 1929,1
“The anatomic structure of the spleen is designed exceedingly well for the two functions previously described, that is, cellular destruction and storage.” 1950,2
Introduction
Hypersplenism refers to a clinical condition in which an enlarged spleen leads to excessive pooling and destruction of blood cells, resulting in cytopenias such as anemia, leukopenia, and thrombocytopenia. Despite the low peripheral blood counts, the bone marrow is typically normal or hypercellular, reflecting intact or even increased production. The pathophysiology involves both sequestration of circulating blood elements and enhanced phagocytic activity within the spleen. Hypersplenism is most commonly seen in the setting of portal hypertension (e.g., from cirrhosis), but can also occur with infiltrative or hematologic disorders. Recognition is important, as the cytopenias are usually well-tolerated but can complicate underlying disease management.
History
Original use of the term
“It is but a step then to assume that there may exist for the spleen conditions associated with an hyperactivity of some of its functions, let us say the function of influencing hemolysis. To such a condition the term “hypersplenism” may be applied . . . If it can be shown that important clinical symptoms consistently point to a hyperfunction of the spleen, and that these symptoms disappear, or are strikingly mitigated, when the spleen is removed from the body, an important step will have been taken toward defining the changes in function of the spleen.” John H. King of Johns Hopkins, reporting in August 1914 on “Studies in the Pathology of the Spleen. According to Doan, “This is the first use of the term “hypersplenism,” which we have found in the medical literature.”3
Evolving definitions
- In the 1930s, the concept of hypersplenism was more formally codified: cytopenias (anemia, leukopenia, thrombocytopenia, or pancytopenia), a hyperplastic bone marrow, splenomegaly, and correction after splenectomy — what became the “classic tetrad” of hypersplenism.
- A 1948 paper offered the following definition: “Hypersplenism is defined as an exaggeration of splenic function resulting in reduction of one or more of the cellular elements of the blood (excessive sequestration and destruction of blood cells).”4
- In the 1940s, Wiliam Damashek defined hypersplenism as an adequate history plus splenomegaly plus anemia, neutropenia, thrombocytopenia or pancytopenia, plus a normal or hyperplastic bone marrow.5
- According to Charles Doan (1949), the diagnosis of a splenic blood dyscrasia rested mainly on five important criteria:6
- Diminution of one or more of the circulating elements of the bloods
- Normal or increased bone marrow activity
- Splenic enlargement
- Increase in the number of deficient blood elements after subcutaneous injection of adrenalin
- Complete rapid clinical recovery of the patient following splenectomy.
- Krache and Riser, 1949:

- Ries CA, 1973:
- Hypersplenism was defined as anemia, leukopenia, thrombocytopenia, or a combination of these resulting from excessive, splenic sequestration or pooling of blood cells, usually associated with clinical splenomegaly and always ameliorated by splenectomy.
- As we will see below, modern usage is more flexible:
- In practice, clinicians often diagnose hypersplenism without the splenectomy “proof,” since we rarely remove spleens just to confirm the diagnosis.
- The core triad most people use today is:
- Enlarged spleen
- Cytopenias
- Marrow showing appropriate/compensatory hematopoiesis
- Splenectomy response is considered supportive evidence rather than a requirement.
- To summarize:
- Strict, historical definition = requires splenectomy response.
- Current, pragmatic definition = splenomegaly + cytopenias + intact marrow response.
Two mechanisms proposed
- The mechanical hypothesis of cellular sequestration, phagocytosis and lysis by the spleen:7
- The spleen causes cytopenias by excessive pooling, sequestration, or both, and ultimate destruction of the circulating blood cells.
- Support for this hypothesis, which was espoused by Doan, and has prevailed to this day:
- Kinetic studies using isotopically labeled blood cells demonstrated increased cell destruction with splenic sequestration, and a compensatory increase in production of the involved cell lines by the bone marrow.
- Direct counts of blood cells from splenic artery and vein demonstrated cell density gradients across the splenic circulation in experimental animals and in patients with hypersplenism.
- The hormonal hypothesis of excessive marrow inhibition by the spleen (spleen as an endocrine organ):
- The spleen produces humoral factors which suppress production or release of blood cells from the bone marrow.
- This hypothesis (espoused by Damashek) fell out of favor because no such factors could be identified, and because the bone marrow of patients with hypersplenism usually shows hyperplasia of the precursors of the cells that are decreased in the peripheral blood.
Types of hypersplenism
- In the mid 1900s, hypersplenism was classified as primary vs. secondary:
- Primary:
- Hereditary spherocytosis (“familial hemolytic ictero-anemia”)
- Idiopathic thrombocytopenic purpura (ITP)
- Splenic neutropenia and panhematopenia
- Acquired hemolytic anemia
- Secondary:
- Developing in the course of other diseases such as:
- Chronic infections
- Autoimmune diseases
- Amyloidosis
- Chronic congestive splenomegaly (e.g., Banti’s syndrome)
- Hematologic malignancies
- Developing in the course of other diseases such as:
- In 1973, Ries wrote: “Hypersplenism resulting from splenic enlargement has traditionally been considered to be either primary or secondary. It is doubtful, however, whether true primary hypersplenism exists. The older terminologies of primary hypersplenism and primary splenic anemia and neutropenia probably reflect a lack of understanding of the true pathogenesis of these disorders. In the absence of demonstrable histopathologic changes in the spleen and in the absence of portal hypertension, the cytopenias previously regarded as primary hypersplenism are likely the result of either intrinsic defects in the blood cells themselves or immune cytopenias in which antibody activity has not been clearly demonstrated.”8
- Primary:

- According to cell lines involved:
- Splenic anemia, congenital hemolytic icterus
- Splenic thrombocytopenia
- Splenic neutropenia
- Splenic panhematopenia
Definition
Hypersplenism is a clinical syndrome characterized by:
- Presence of splenomegaly.
- Peripheral cytopenias (anemia, leukopenia, and/or thrombocytopenia).
- Normal or hyperplastic bone marrow (showing that the marrow is capable of producing blood cells).
- Improvement of cytopenias after splenectomy (the diagnostic “proof”).
The classic “hypersplenism” definition includes the “reversal after splenectomy” criterion, but that’s retrospective and not very useful at the bedside when you haven’t (and usually won’t) remove the spleen. So, in practice, hypersplenism is defined before splenectomy as:
- Presence of splenomegaly
- Peripheral cytopenias (affecting one or more lineages)
- Bone marrow showing normal or increased cellularity (i.e., preserved or even up-regulated production, excluding primary marrow failure)
- No other explanation for the cytopenias
Splenectomy response is sometimes listed as a confirmatory feature but not required for the working definition in a living patient.
Note: Hypersplenism is not a primary disease, but rather a syndrome that develops in the setting of splenomegaly from diverse causes.
Pathophysiology
The underlying mechanism is increased pooling/sequestration and/or destruction of blood cells in the enlarged spleen, sometimes combined with altered regulation of production (e.g., via thrombopoietin for platelets).
- Mechanisms of hypersplenism (each of the following takes place in the splenic cords of the red pulp):
- Pooling/sequestration:
- In splenomegaly, the spleen becomes a large reservoir for circulating blood cells.
- Normally, ~30% of platelets are sequestered in the spleen; in hypersplenism this may rise to 80–90%.
- Red cells and neutrophils can also be held back, leading to pancytopenia.
- Increased destruction/phagocytosis (red pulp):
- The enlarged spleen has more macrophages and heightened reticuloendothelial activity.
- Trapped cells are more likely to be phagocytosed and destroyed.
- This contributes to the shortened lifespan of red cells, platelets, and leukocytes.
- Pooling/sequestration:
- Key Points:
- Sequestration explains why cells are diverted away from circulation.
- Destruction explains why they don’t simply re-enter the blood but are instead prematurely removed.
- Hypersplenism is usually a combination of pooling and accelerated clearance, which together produce cytopenias — with the marrow often responding by ramping up production.
Pathophysiology of Hypersplenism: Pooling vs. Destruction
| Mechanism | Process | Predominant Cell Effect | Clinical Examples | Can cause hypersplenism |
|---|---|---|---|---|
| Pooling (Sequestration) Alone | Enlarged spleen traps increased proportion of circulating cells in cords/sinusoids; cells remain viable but unavailable | Platelets, neutrophils (short lifespan, regulated by circulating mass) | Portal hypertension (cirrhosis), splenic vein thrombosis, congestive splenomegaly | Yes |
| Increased Destruction Alone | Hyperactive macrophages phagocytose blood cells prematurely | Red cells (compensation less effective if destruction is brisk) | Autoimmune hemolytic anemia, ITP | Typically no because spleen size normal |
| Mixed (Pooling + Destruction) | Sequestration slows flow, facilitating phagocytosis | All three lineages can be affected | Congenital hemolytic anemias (HS, HE, thalassemia, sickle cell, enzyme deficiencies), MPN, malaria, visceral leishmaniasis | Yes |
A note about white vs. red pulp
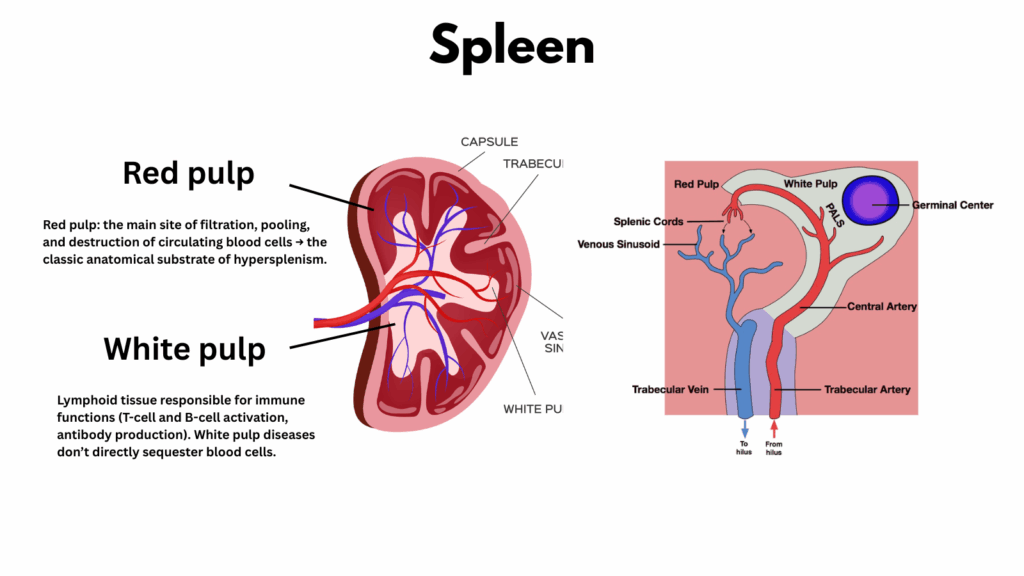
The spleen is divided into two functional compartments: the white pulp and the red pulp. The white pulp consists of islands of lymphoid tissue surrounding arterioles and serves as the immune surveillance center of the spleen. The red pulp (which comprises about 75% of the splenic volume), in contrast, is made up of splenic cords and sinusoids lined with macrophages; it functions as the body’s filtration and recycling system, removing old or damaged red blood cells and salvaging useful components such as iron. Together, the white and red pulp make the spleen both an immune sentinel and a quality-control organ for the blood.
Splenomegaly/hypersplenism mainly reflects expansion of the red pulp (blood filtration work increases).
- Red pulp = Filtration & Recycling Center:
- The main site of filtration, pooling, and destruction of circulating blood cells → the classic anatomical substrate of hypersplenism.
- Like a quality-control factory line. Every RBC must squeeze through narrow slits; those that fail are removed.
- White pulp = = Immune Surveillance Hub
- Lymphoid tissue responsible for immune functions (T-cell and B-cell activation, antibody production).
- Diseases that cause splenomegaly by expanding the white pulp (e.g., lymphomas, CLL, hairy cell leukemia, chronic infections like malaria or kala-azar) don’t directly sequester blood cells.
- Instead, they cause splenomegaly + immune activation.9.
Patterns of cytopenias
- Thrombocytopenia is the most frequent manifestation, since platelets are most sensitive to splenic sequestration.
- Leukopenia (especially neutropenia) is less common.
- Anemia is uncommon purely from hypersplenism (usually multifactorial if present).
Causes
Because the terms splenomegaly and hypersplenism are often used interchangeably, it is important to distinguish between them. Splenomegaly simply refers to an enlarged spleen, regardless of whether blood counts are affected. Hypersplenism, on the other hand, describes a functional syndrome in which an enlarged spleen causes cytopenias through pooling and/or destruction of blood cells. Not all causes of splenomegaly lead to hypersplenism, and not all cytopenias due to splenic destruction represent hypersplenism unless the spleen is enlarged. To make this distinction clear, it is useful to consider the causes of splenomegaly separately from the causes of hypersplenism.
- Splenomegaly:
- Major categories of causes:
- Congestive – e.g., portal hypertension (cirrhosis, splenic vein thrombosis, right heart failure).
- Infectious/immune stimulation – e.g., EBV, CMV, malaria, HIV.
- Hematologic malignancy – e.g., leukemias, lymphomas, myeloproliferative neoplasms.
- Infiltrative/storage – e.g., Gaucher, Niemann-Pick, amyloidosis, sarcoidosis.
- Autoimmune
- Others – trauma/hematoma, cysts.
- Major categories of causes:
- Hypersplenism:
- Pooling – congestive splenomegaly (cirrhosis/portal hypertension).
- Pooling + destruction – congenital hemolytic anemias, malaria, visceral leishmaniasis, myeloproliferative neoplasms.
- Destruction alone (e.g., autoimmune hemolytic anemia with a normal-sized spleen) causes cytopenias but is not hypersplenism because splenomegaly is absent.
Do all causes of splenomegaly result in hypersplenism?
- No, some causes of splenomegaly enlarge the spleen without significant cytopenias.
- Splenomegaly that often → hypersplenism:
- Congestive: portal hypertension, hepatic vein/portal vein thrombosis → pooling.
- Work hypertrophy: hemolysis (HS, thalassemia, sickle), infections (malaria, kala-azar, EBV), immune stimulation.
- Infiltrative/MPN: myelofibrosis, hairy cell leukemia, Gaucher disease.
- These enlarge the red pulp or both red + white pulp, creating sequestration and destruction → cytopenias.
- Splenomegaly that does not always cause hypersplenism:
- Isolated splenic cysts, hemangiomas, benign tumors → can enlarge spleen without changing filtering activity.
- Some indolent lymphomas: spleen can be big but not always associated with cytopenias.
- Early portal hypertension: may enlarge spleen before cytopenias appear.
- Congenital “wandering spleen” or splenic ptosis: large/abnormally located but not functionally overactive.
- Key teaching point:
- All hypersplenism requires splenomegaly.
- But not all splenomegaly progresses to hypersplenism.
- It depends on whether the enlargement is accompanied by functional overactivity of sequestration and destruction.
| Cause of Splenomegaly | Causes Hypersplenism? | Mechanism | Comment |
|---|---|---|---|
| Congestive | Yes | Pooling | Pooling. The most classic cause of hypersplenism |
| Work Hypertrophy | Yes | Overactive clearance | Increased macrophage/RES activity |
| Infiltrative | Variable | Expansion of pulp crowds out normal function and increases pooling | Hypersplenism is common in myelofibrosis, hairy cell leukemia, Gaucher disease, but not inevitable in all infiltrative causes |
| Immune/Inflammatory Hyperplasia | Sometimes | Lymphoid hyperplasia and reticuloendothelial stimulation → increased clearance. | Hypersplenism is especially common in malaria, kala-azar, tropical splenomegaly syndrome, less so in transient viral infections. |
| Neoplastic | Rare | Usually cause splenomegaly without significant hypersplenism, unless very bulky or associated with secondary congestion. | Primary splenic tumors (hemangioma, angiosarcoma, lymphoma, metastases). |
| Cystic | No | N/A | N/A |
A Word About Portal Hypertension
Among the many causes of hypersplenism, portal hypertension deserves special attention. It is by far the most common setting in which hypersplenism arises, particularly in patients with cirrhosis. The mechanism is primarily pooling, namely congestion of splenic blood flow causes the spleen to enlarge and sequester platelets and leukocytes, often leading to cytopenias even in the absence of increased destruction. In addition, cirrhosis is associated with reduced hepatic production of thrombopoietin (TPO), further compounding thrombocytopenia. Because portal hypertension is so prevalent worldwide, understanding its role in hypersplenism is critical for clinicians: it explains why cytopenias are frequent in cirrhosis, influences management decisions, and highlights how both vascular physiology and cytokine regulation of platelet mass underlie this clinical syndrome.
- Pathophysiology:
- Portal hypertension (from cirrhosis, non-cirrhotic portal fibrosis, schistosomiasis, etc.) → increased splenic venous pressure → congestive splenomegaly.
- The enlarged spleen sequesters (and ultimately destroys) more blood cells → hypersplenism.
- Both pooling (expanded splenic reservoir) and accelerated destruction (phagocytosis in the red pulp) contribute.
- Platelets are most sensitive because:
- Splenic sequestration:
- Enlarged spleen traps a disproportionate fraction of platelets (up to 80–90% vs ~30% normally).
- Short lifespan (~7 days). “Time out” from circulation is almost equivalent to destruction.
- Regulated by thrombopoietin in relation to total mass (circulating + splenic), so sequestration doesn’t trigger increased TPO → no compensatory marrow drive.
- Thrombopoietin (TPO) biology:
- TPO is produced constitutively by the liver.
- Circulating platelet mass determines how much TPO is cleared:
- Low platelet mass → high free TPO → stimulates megakaryocytes.
- High platelet mass (even if many are splenic) → low free TPO → less marrow stimulation.
- In portal hypertension, two hits combine:
- Increased platelet mass in the spleen → more TPO bound and removed from circulation.
- Reduced hepatic TPO production in cirrhosis → less baseline stimulus for megakaryopoiesis.
- So the marrow sees low TPO despite low circulating counts → poor compensation.
- Splenic sequestration:
- Epidemiology:
- Hypersplenism is very common in cirrhosis with portal hypertension.
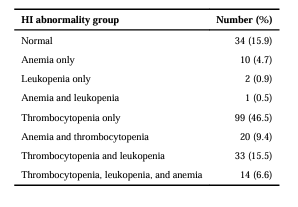
- Thrombocytopenia occurs in up to 70–90% of patients with cirrhosis and splenomegaly.
- Leukopenia (~30–50%) and anemia (~10–20%) are less frequent, and usually multifactorial.
- Clinical features:
- Splenomegaly on exam or imaging.
- Thrombocytopenia: often the first and most prominent cytopenia.
- Leukopenia/neutropenia: contributes to infection risk.
- Anemia: typically mild unless other contributors (GI bleeding, marrow suppression from alcohol/virus, reduced EPO in renal impairment, nutritional deficiencies).
- Diagnosis:
- Suspected when a patient with cirrhosis + splenomegaly has cytopenias with:
- Preserved or hypercellular bone marrow.
- No other obvious cause of cytopenias.
- Reticulocytes often normal or slightly elevated (depending on bleeding/hemolysis).
- Exclude other contributors: marrow suppression (alcohol, hepatitis C), nutritional deficiency, drug effects.
- Suspected when a patient with cirrhosis + splenomegaly has cytopenias with:
- Clinical importance:
- Thrombocytopenia complicates:
- Liver biopsy, variceal banding, surgery, or other invasive procedures.
- Administration of certain antivirals or chemotherapy.
- Cytopenias may mask marrow pathology in cirrhotic patients.
- Thrombocytopenia complicates:
- Management:
- Treat the underlying portal hypertension:
- Antiviral therapy for hepatitis.
- Abstinence in alcohol-related cirrhosis.
- TIPS (transjugular intrahepatic portosystemic shunt) can decompress portal hypertension and reduce splenomegaly.
- Splenectomy (or less commonly, splenic artery embolization) can correct cytopenias, but carries risks (bleeding, infection, thrombosis). Reserved for select cases.
- Supportive strategies: platelet transfusion, TPO receptor agonists to raise platelets before procedures.
- Treat the underlying portal hypertension:
Does hypersplenism require splenomegaly?
Does hypersplenism require splenomegaly? The short answer is yes. By definition, hypersplenism is the functional consequence of an enlarged spleen. Cytopenias that occur in the absence of splenomegaly—such as those caused by immune-mediated destruction—do not qualify as hypersplenism. This may sound like splitting hairs, but the distinction is so important that it bears repetition and some unpacking: splenomegaly is a structural finding, while hypersplenism is the functional syndrome that arises when an enlarged spleen lowers circulating blood counts through pooling and/or destruction.
- By classic definition, hypersplenism requires splenomegaly.
- The “syndrome of hypersplenism” is usually defined by four criteria:
- Splenomegaly
- One or more cytopenias (anemia, leukopenia, thrombocytopenia, pancytopenia)
- Normocellular or hypercellular marrow (showing intact production)
- Improvement after splenectomy
- The “syndrome of hypersplenism” is usually defined by four criteria:
- The rationale is that the enlarged spleen provides both the physical space (increased red pulp volume) and heightened reticuloendothelial activity that allow for abnormal sequestration and destruction of blood cells.
- Without splenomegaly, sequestration and destruction can still occur (for example in some autoimmune cytopenias where the spleen removes opsonized cells), but this would not traditionally be called “hypersplenism.” Instead, it would be described as splenic sequestration or simply part of the disease mechanism (e.g., autoimmune hemolysis, ITP). If the spleen is normal in size, other mechanisms of cell destruction or removal are at play, but it doesn’t meet the formal definition of hypersplenism.
Why don’t ITP or AIHA cause hypersplenism, whereas congenital hemolytic anemias do?
When discussing cytopenias related to the spleen, it is important to distinguish immune-mediated destruction (as in autoimmune hemolytic anemia and ITP) from the mixed mechanism seen in congenital hemolytic anemias. In the former, splenomegaly is uncommon because the spleen removes only those cells that have been specifically tagged by antibodies, a highly targeted process. By contrast, in congenital hemolytic anemias, red cells are mechanically abnormal and get trapped in the splenic cords, where they are indiscriminately removed, leading to both pooling and destruction and, ultimately, splenomegaly. This difference explains why autoimmune cytopenias are not hypersplenism, while congenital hemolytic anemias are classic examples.
- In autoimmune cytopenias (ITP, AIHA), splenomegaly is uncommon:
- The cords themselves aren’t packed or congested; overall spleen architecture is normal.
- antibodies coat the blood cells (IgG on platelets in ITP, IgG/C3 on RBCs in AIHA
- Splenic macrophages in the red pulp recognize these opsonized cells via Fc and complement receptors and clear them. The cytopenias are due to immune-mediated clearance of platelets, not architectural expansion.
- Analogy: This is the “smart bomb” model: macrophages don’t increase in number or size, they just selectively remove the cells that have been tagged. The rest of the traffic flows freely.
- Result: cytopenias without splenomegaly → not hypersplenism.
- In congenital hemolytic anemias (HS, HE, thalassemia, sickle cell, enzyme defects, splenomegaly is typical:
- The abnormal RBCs have altered deformability, so they cannot squeeze through the splenic cords and sinusoids efficiently. They get mechanically held up.
- Because so many cells are stalled, the spleen expands with congested red pulp (pooling), and resident macrophages “pick off” these delayed or deformed cells (destruction).
- Analogy: This is less like smart bombs and more like a minefield or cluster bomb — there’s no precision targeting; instead, any cell that stumbles or lingers in the danger zone gets destroyed.
- Result: Both pooling and destruction → splenomegaly with cytopenias → hypersplenism.
- Response to splenectomy:
- In hypersplenism, splenectomy corrects cytopenias by removing the enlarged, overactive organ.
- In ITP, splenectomy works not because of hypersplenism but because it removes the site of autoantibody-mediated clearance (and some antibody production).
Not all blood cells are created equal
When it comes to hypersplenism, red cells, platelets, and white cells do not share the same fate. Each lineage has a distinct “journey” through the spleen and a different mechanism by which counts fall when the organ enlarges. Red cells are subjected to a rigorous deformability test in the splenic cords, platelets are passively pooled within the expanded splenic reservoir, and white cells are slowed and marginated in the low-flow environment of the congested red pulp. Drilling down on these differences highlights that hypersplenism is not a single process, but a convergence of three lineage-specific mechanisms.
- Red cells:
- The “quality control gauntlet” in the splenic cords and sinusoidal slits is primarily about RBCs.
- Only deformable, biconcave RBCs can slip through; less deformable ones (aged, spherocytic, sickled, parasitized) get hung up and then cleared by macrophages.
- For RBCs, hypersplenism results from pooling due to reduced efficiency of mechanical filtration, coupled with increased phagocytosis of the trapped cells.
- Platelets:
- Platelets are much smaller and usually pass through sinusoids without trouble.
- Their sequestration in hypersplenism is less about mechanical trapping and more about volume dynamics:
- In normal physiology, ~30% of platelets are “parked” in the spleen.
- When the spleen enlarges (especially with red pulp congestion/expansion), the splenic vascular bed and cords become larger, and more platelets are passively held there.
- It’s not that platelets “can’t get through,” but rather that the expanded reservoir soaks them up.
- Destruction of platelets is not a big feature unless there’s an immune overlay.
- So, platelet hypersplenism = pooling in an enlarged sponge, not filtration failure.
- White blood cells:
- Under normal conditions, most WBCs take the closed circulation shortcut (arteriole → venule) and avoid the cords.
- They mainly enter sinusoids via the closed circulation.10he open vs. closed circulation are two entrances, but they both converge on the same sinusoidal channels that ultimately drain to venules and the splenic vein.
- Once inside sinusoids, in normal flow, they keep moving. In splenic congestion, flow slows. This sluggish, low-shear environment increases the probability that circulating leukocytes will marginate (i.e., move toward and transiently adhere to the sinusoidal endothelium).
- Summary:
- RBC cytopenia = open circulation gauntlet (cords → sinusoidal slits).
- Platelet cytopenia = pooling in enlarged vascular reservoir.
- WBC cytopenia = margination/sequestration within sinusoids reached via closed circulation.
Is pooling and destruction of cells necessary to cause hypersplenism-associated cytopenias?
- Red blood cells:
- If hypersplenism were only pooling (without destruction):
- RBCs would be sequestered in the spleen, so the circulating RBC count/hematocrit would drop.
- That means renal oxygen sensing would see reduced oxygen delivery → trigger ↑ EPO → bone marrow would ramp up production.
- The spleen would then release some of its sequestered cells back into circulation (since sequestration is dynamic, not permanent).
- Net effect: the marrow could compensate, and steady-state anemia would not develop.
- Thus, for anemia to develop, need:
- Splenic destruction/shortened RBC lifespan:
- Trapped RBCs undergo mechanical stress and prolonged contact with macrophages → accelerated clearance.
- Marrow can’t keep up → anemia develops.
- Impairment of the oxygen-sensing → EPO → erythropoiesis axis
- Chronic kidney disease → ↓ EPO production.
- Bone marrow disease (MDS, aplasia, fibrosis, infiltration) → impaired erythropoietic response.
- Nutritional deficiencies (iron, folate, B12) → limit marrow’s ability to respond.
- Combined mechanisms
- For example, cirrhosis with portal hypertension:
- Splenomegaly → sequestration + destruction.
- Concurrent marrow suppression from alcohol, hepatitis C, or chronic disease → blunted compensatory response.
- For example, cirrhosis with portal hypertension:
- Splenic destruction/shortened RBC lifespan:
- Teaching pearl:
- Pooling alone ≠ anemia (healthy marrow + intact EPO can compensate).
- Anemia in hypersplenism = pooling + destruction, or pooling + impaired marrow/EPO response.
- If hypersplenism were only pooling (without destruction):
- Platelets:
- Unlike RBCs, platelets have a very short lifespan (≈ 7–10 days).
- In splenomegaly, as much as 80–90% of the platelet mass can be sequestered in the spleen at any given time.
- Even if not actively destroyed, those platelets are effectively “out of circulation.”
- Platelets → Regulated by total platelet mass (via TPO feedback)
- The liver makes thrombopoietin (TPO) at a mostly constant rate.
- Platelets bind and clear TPO from plasma.
- Bioavailable TPO is inversely proportional to the total platelet mass (whether platelets are in circulation or sequestered in the spleen).
- So in hypersplenism, even if most platelets are sequestered in the spleen, they still bind TPO.
- Marrow “sees” low circulating counts but doesn’t increase production much, because total platelet mass is normal.
- Result: thrombocytopenia persists even though the marrow could make more.
- So, do platelets need destruction too?
- Not necessarily.
- Pooling alone can lower the circulating platelet count to the point of thrombocytopenia.
- Sequestration does not trigger the same marrow “alarm” signal that low circulating platelets would, since TPO sensing is mass-based.
- In practice, though, both mechanisms contribute:
- Sequestration into an enlarged red pulp.
- Enhanced phagocytosis by splenic macrophages (especially if platelets are antibody-coated, as in ITP).
- Not necessarily.
- White blood cells:
- Neutrophils: half-life ≈ 6–10 hours in blood
- Regulation: by demand-driven cytokines (G-CSF, GM-CSF, M-CSF, interleukins).
- Signal: infection, inflammation, or stress → ↑ colony-stimulating factors → ↑ granulopoiesis/monocytopoiesis.
- Unlike platelets, there is no mass-based sensing system.
- Hypersplenism effect: sequestration + destruction lower circulating neutrophils/monocytes/lymphocytes. The marrow may become hypercellular, but unless there is infection/inflammation, there is no strong cytokine signal to “overproduce.” Thus leukopenia persists.
- Like platelets, pooling alone is enough for leukopenia to occur in hypersplenism
- Summary:
- RBCs: EPO system = oxygen-based → protects against sequestration-only anemia.
- Platelets: TPO system = mass-based → sequestration looks “normal,” so thrombocytopenia persists.
- WBCs: cytokine demand-based → without infection/inflammation, marrow doesn’t fully compensate for splenic pooling/destruction.
- Teaching pearl:
- The shorter a cell’s lifespan, the more likely sequestration alone will lower its circulating count, because many sequestered cells will die before rejoining circulation.
A visual tour of splenomegaly and hypersplenism
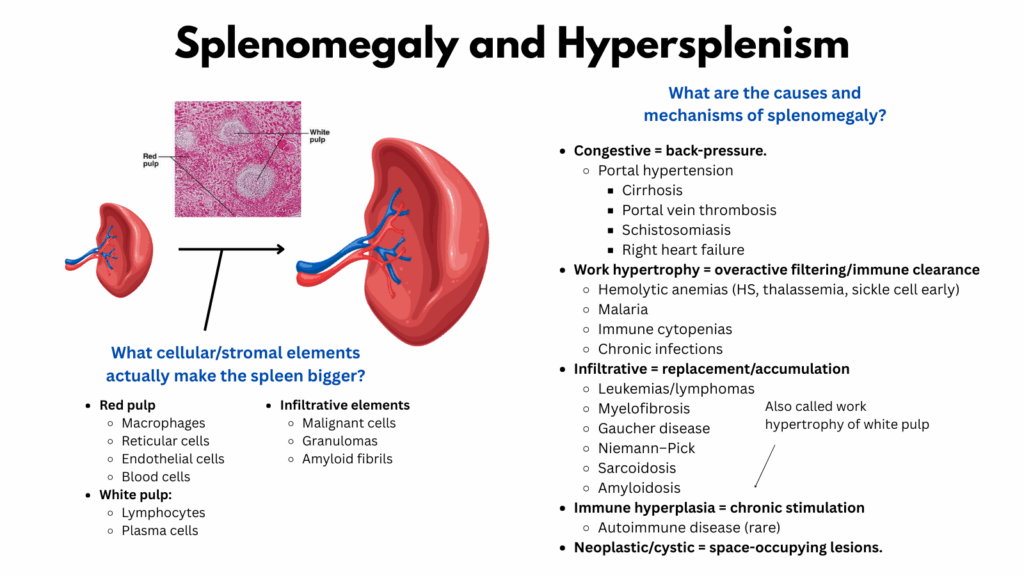
This image illustrates splenomegaly, or an enlarged spleen. Between the two spleens is a histology section showing white pulp and red pulp; the red pulp makes up about 75% of the spleen’s total volume. Functionally, the red pulp serves as the filtration and recycling center, where blood cells (especially red blood cells) must pass quality control in the splenic cords. The white pulp, by contrast, is an immune surveillance hub composed of lymphoid tissue.
On the right side of the slide is a classification of causes and mechanisms of splenomegaly. They can be grouped into:
- Congestive splenomegaly, typically due to portal hypertension.
- Work hypertrophy, which most often reflects increased filtering activity in the red pulp, but can also involve expansion of the white pulp.
- Infiltrative causes, where cells or extracellular material infiltrate and replace normal splenic tissue.
- Miscellaneous causes, which don’t fit neatly into the other categories.
So what actually enlarges the spleen? Only a few things can contribute to increased splenic volume: cells and extracellular material.
- In the red pulp, macrophages (engulfing red cells), reticular cells (providing structural framework), endothelial cells (lining arterioles and sinusoids), and pooled blood cells themselves all add to bulk. Platelet pooling, for instance, can be significant.
- In the white pulp, lymphocytes and plasma cells may expand.
- Among infiltrative elements, malignant cells, granulomas, and amyloid fibrils can accumulate.
Understanding splenomegaly is essential because it sets the stage for hypersplenism. By definition, hypersplenism requires an enlarged spleen, and some causes of splenomegaly are much more likely than others to lead to hypersplenism (for example, congestive splenomegaly is more often associated than infiltrative splenomegaly).
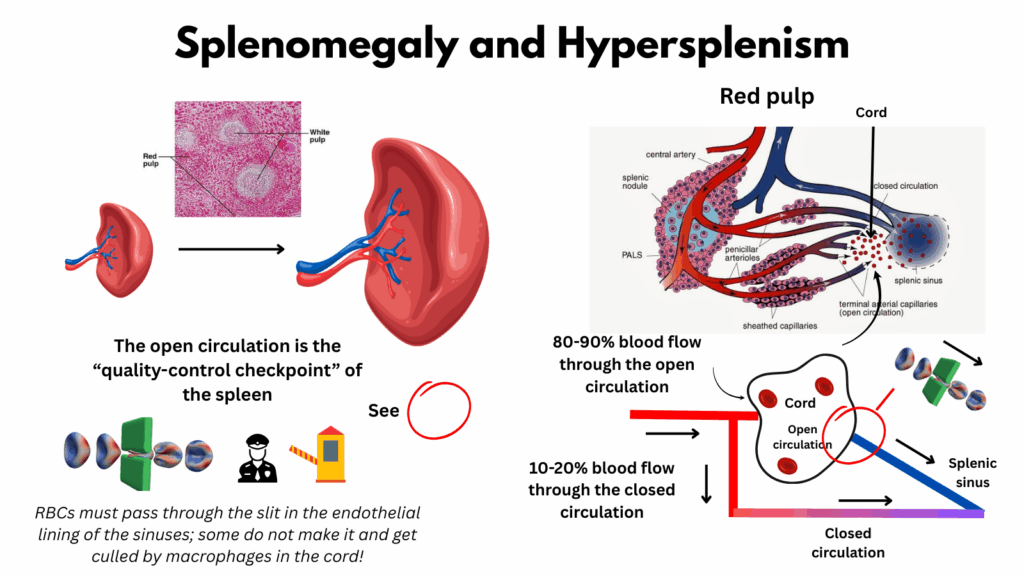
In this image, we zoom in on the splenic cords (or cords of Billroth). The name can be misleading; it suggests a solid structure, but in reality the cords are open spaces, part of the spleen’s open circulation. Here, blood cells pool while awaiting their chance to exit through the extremely narrow slits between endothelial cells of the adjacent splenic sinusoids.
On the right (top panel), the diagram shows the red pulp vasculature:
- Arteries (red for oxygenated blood) branch into terminal arterial capillaries.
- From these, red blood cells (depicted as red circles) spill out into a large open space—the cords—before attempting to enter the splenic sinus on the right, shown in blue to indicate venous, deoxygenated blood.
- Importantly, some arterial blood flows directly into the sinus without ever leaving the vessel. This is the closed circulation, which accounts for only ~10–20% of splenic blood flow. For the remaining majority, cells must endure the “purgatory” of the open cords before reaching the sinusoids.
The simplified diagram below illustrates the same concept: blood entering the red pulp may either:
- Exit into the cords (open circulation), or
- Remain within vessels (closed circulation).
In both pathways, blood ultimately drains into the venous sinus, and then the splenic vein.
In the bottom-left illustration, the focus shifts to the quality control challenge red cells face. To re-enter circulation, they must squeeze through the tight endothelial slits of the sinus walls. Normal, deformable red cells can pass; damaged or rigid cells cannot and are removed by macrophages that patrol the cords, like birds of prey waiting for vulnerable hatchlings, picking off those that fail to make it safely to the “ocean” of the venous system.
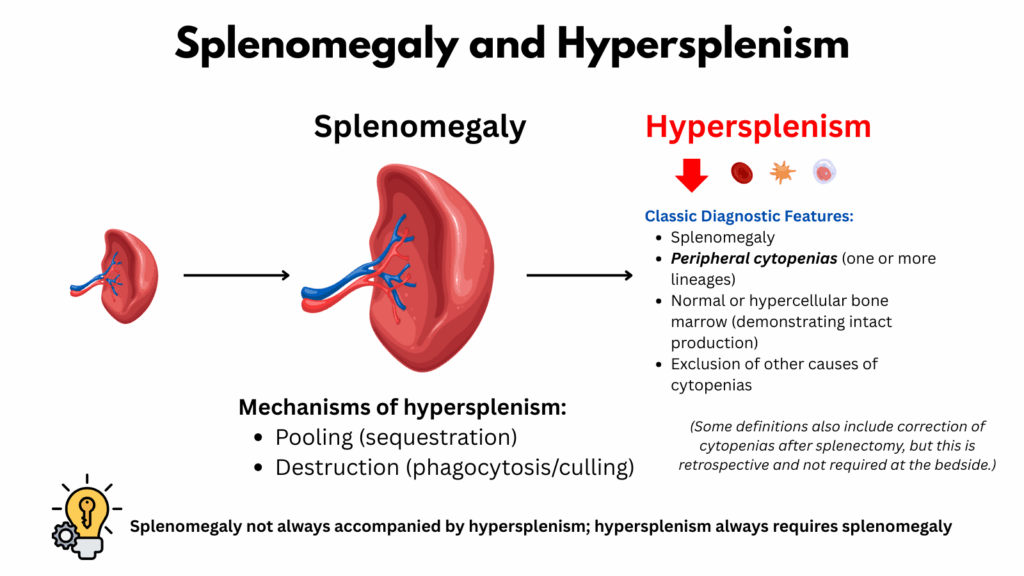
This slide defines hypersplenism and introduces its mechanisms.
Let’s start with the definition. The classic diagnostic features include:
- Splenomegaly
- Peripheral cytopenias affecting one or more lineages
- A normal or hypercellular bone marrow
- Exclusion of other causes of cytopenias
Earlier definitions also required correction of cytopenias after splenectomy. That criterion is no longer practical. Splenectomy is uncommon today, and using it as proof makes hypersplenism a retrospective diagnosis. In reality, we need to be able to make the diagnosis before considering splenectomy.
It is also important to note the distinction between splenomegaly and hypersplenism.
- Splenomegaly is a structural/anatomical condition that refers to the physical enlargement of the spleen.
- Hypersplenism is a syndrome or complication that arises from splenomegaly and is characterized by cytopenias.
- Not all splenomegaly results in hypersplenism, but hypersplenism always requires splenomegaly.
Now to the mechanisms. There are two principal processes:
- Pooling or sequestration of blood cells – primarily in the cords of Billroth.
- Destruction (phagocytosis) of blood cells – carried out by macrophages in the cords, leading to their clearance from circulation.
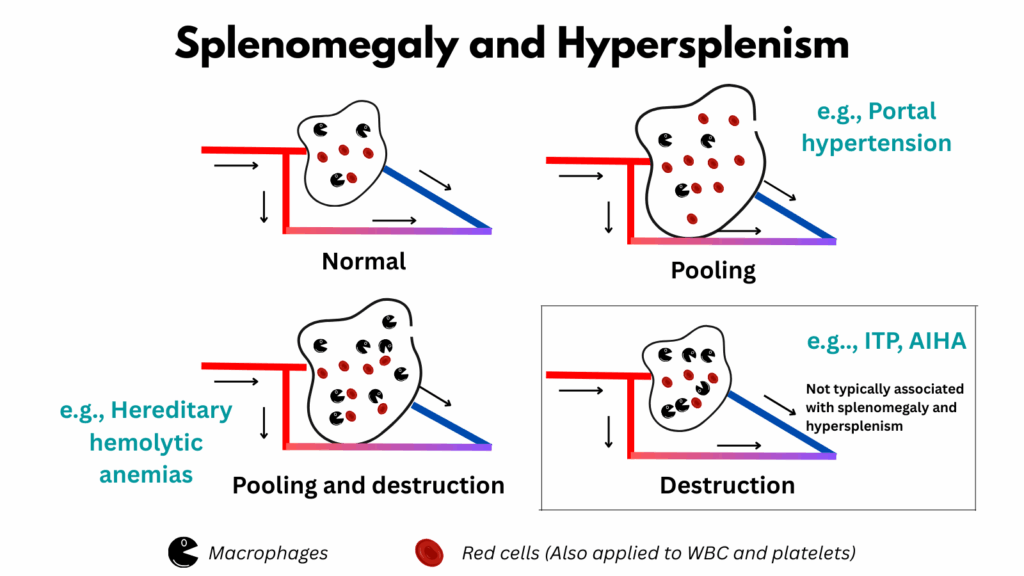
This image highlights the mechanisms of hypersplenism: pooling and destruction.
- Pooling (upper right):
Pooling is a passive process in which blood cells accumulate within the splenic cords, leading to expansion of the red pulp. - Destruction (lower right):
Destruction refers to increased phagocytosis of blood cells by macrophages, shown here as Pac-Man icons. Importantly, destruction alone does not necessarily produce splenomegaly. For example, in immune thrombocytopenia (ITP), macrophages specifically target antibody-coated platelets. Other blood cells pass freely into the venous sinuses, so there is no need for splenic enlargement. Thus, cytopenias (thrombocytopenia in ITP, anemia in autoimmune hemolytic anemia, or both in Evans syndrome) can occur without hypersplenism. - Pooling + Destruction (lower left):
In some conditions, both processes occur together. Here, the problem is not pressure backup from portal hypertension, but rather the inability of abnormal red cells to escape from the cords into the sinusoids. This leads to both a backlog of cells (pooling) and increased macrophage activity (destruction). A classic example is seen in hemolytic anemias.