Hemostasis is a spatially organized, threshold-dependent system; bleeding phenotype reveals which structural layer has failed.
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
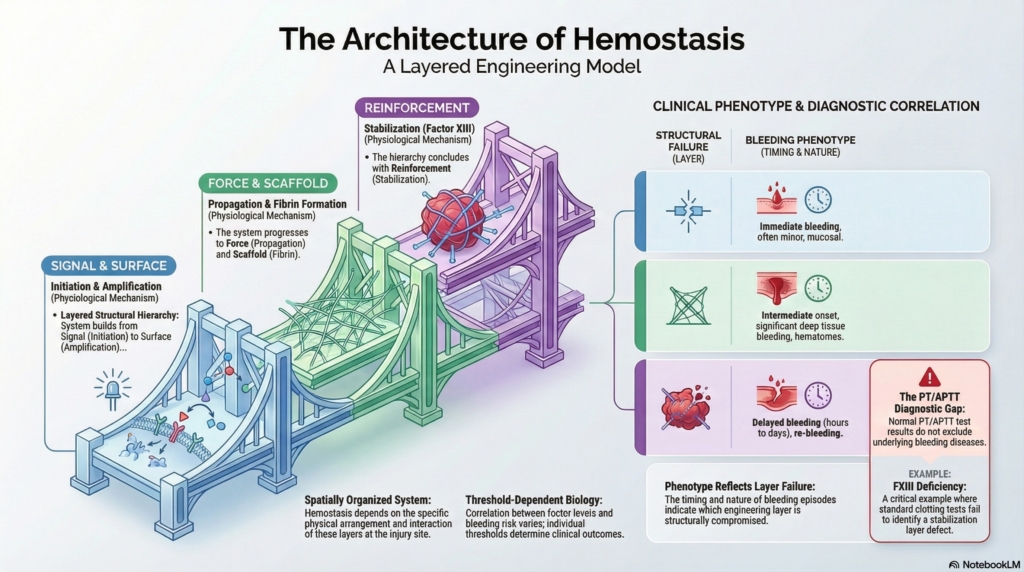
Hemostasis as a layered biological system
Hemostasis is not a simple linear cascade. It is a coordinated, surface-dependent process in which pro- and anticoagulant forces interact on specific cellular and structural platforms.
A useful way to understand this system is as layered structural engineering:
- initiation provides the signal
- amplification builds the surface
- propagation generates force
- fibrin creates the scaffold
- stabilization reinforces the structure
Each layer serves a distinct function. Each failure produces a distinct phenotype.
These processes are spatially organized. Initiation begins on tissue factor–bearing cells. Propagation occurs on activated platelet surfaces. Stabilization occurs within the fibrin scaffold itself.
Qualitative or quantitative defects in any component disrupt this system and produce wide variability in bleeding phenotype, even within the same disorder.
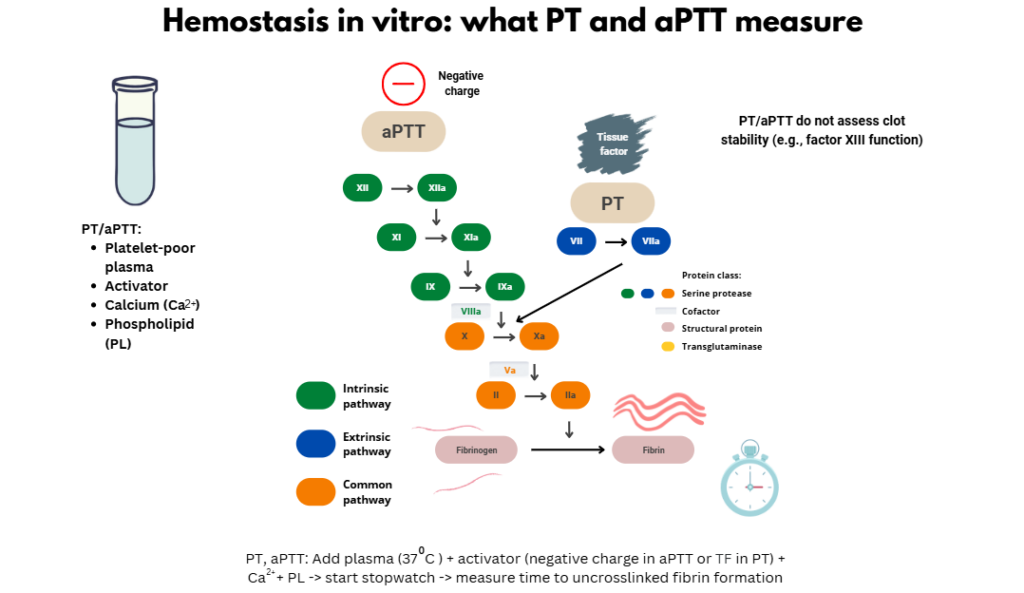
Cascade versus cell-based models
The cascade model remains essential for interpreting laboratory tests:
- PT evaluates factors II, V, VII, and X
- APTT evaluates factors II, V, VIII, IX, X, and XI
- combined prolongation suggests common pathway or fibrinogen defects
However, these tests capture only part of the system. They assess thrombin generation under artificial conditions and do not evaluate clot structure or durability.
They may be normal in clinically significant disorders, including:
- qualitative fibrinogen disorders
- factor XIII deficiency
The cell-based model explains this gap. Coagulation is surface-dependent, thrombin generation is localized, and final clot integrity depends on processes that occur after fibrin is formed.
The cascade explains laboratory patterns.
The cell-based model explains clinical phenotype.
Why phenotype is the primary diagnostic signal
Investigation of bleeding disorders begins with clinical context, not isolated laboratory values. A structured approach considers:
- personal and family bleeding history
- timing of bleeding relative to challenge
- pretest probability of a disorder
- laboratory findings interpreted in context
Timing is particularly informative:
- bleeding at the time of challenge suggests primary hemostasis or early coagulation defects (e.g., platelet disorders, von Willebrand disease)
- bleeding after initial control suggests propagation defects
- bleeding delayed by hours to days suggests defects in stabilization or fibrinolysis
Delayed bleeding is less common and often points to disorders not detected by routine screening.
Phenotype generates the diagnostic hypothesis. Testing confirms it.
Linking mechanism to clinical patterns
Each functional objective of hemostasis produces a characteristic clinical and laboratory pattern:
- initiation defects → abnormal PT, variable phenotype (e.g., factor VII deficiency)
- propagation defects → abnormal APTT, often procedure-dependent bleeding (e.g., factor XI deficiency)
- fibrin defects → abnormal PT and APTT, impaired clot formation (fibrinogen disorders)
- stabilization defects → normal PT and APTT, delayed bleeding (factor XIII deficiency)
- fibrinolysis defects → normal PT and APTT, recurrent late bleeding after initial hemostasis
These patterns reflect where the system fails, not simply which factor is missing.
Initiation: early thrombin and weak phenotype correlation
Factor VII deficiency produces an isolated prolongation of the PT with a normal APTT and highly variable bleeding phenotype.
Large registry studies demonstrate a weak correlation between factor VII activity and clinical severity. Initiation generates an early thrombin signal but does not determine final clot strength. Downstream propagation may compensate, contributing to the disconnect between laboratory abnormality and phenotype.
Propagation: the thrombin burst defines structure
Propagation is driven by tenase and prothrombinase complexes assembled on activated platelet surfaces, producing the thrombin burst required for stable fibrin formation.
Registry data reveal a fundamental distinction. Some factors, such as fibrinogen, factor X, and factor XIII, directly determine clot structure and correlate strongly with bleeding severity. Others, including factor VII and factor XI, operate earlier in the process and show weak or inconsistent correlation with phenotype.
Factor XI deficiency illustrates this principle. Although the APTT is prolonged, bleeding is often procedure-dependent and poorly predicted by factor level. Bleeding tends to occur in tissues with high fibrinolytic activity, particularly mucosal sites such as the oropharynx and genitourinary tract, and is more common in certain populations, including individuals of Ashkenazi Jewish descent.
Propagation behaves as a threshold process. Once thrombin generation exceeds a critical level, fibrin formation becomes robust and relatively insensitive to small changes. Below that threshold, small reductions in activity can produce disproportionate clinical effects.
Fibrin formation: substrate determines architecture
Fibrinogen disorders include both quantitative and qualitative defects, each with distinct clinical implications.
Afibrinogenemia is characterized by markedly prolonged PT and APTT, absent clot formation, and severe bleeding from birth. Diagnosis requires both functional and antigen assays to distinguish quantitative from qualitative abnormalities.
Therapeutic thresholds are mechanistically meaningful. Fibrinogen levels of approximately 1 g/L are typically sufficient for minor bleeding, and 1.5 g/L for major bleeding or procedures. These targets are based on expert consensus and clinical experience rather than randomized trial data.
Below these levels, fibrin structure is insufficient regardless of thrombin generation.
Stabilization: why clots fail after they form
Factor XIII crosslinks fibrin and incorporates antifibrinolytic proteins into the clot, providing structural reinforcement.
Factor XIII deficiency presents with:
- normal PT and APTT
- delayed bleeding
- umbilical stump bleeding
- intracranial hemorrhage
- impaired wound healing
All standard screening tests may be normal. Diagnosis requires a quantitative factor XIII activity assay, as clot solubility tests detect only severe deficiency and are not standardized.
There is a strong correlation between factor XIII activity and bleeding severity. This is a failure of structural reinforcement, not clot formation.
Fibrinolysis: when clots dissolve too soon
Defects in antifibrinolytic pathways, such as α2-antiplasmin or PAI-1 deficiency, produce:
- delayed bleeding
- normal PT and APTT
- normal fibrinogen
The key clinical feature is recurrent bleeding after initial hemostasis.
This pattern differs from factor XIII deficiency. In FXIII deficiency, the clot lacks structural reinforcement. In fibrinolytic disorders, the clot forms and is initially stable but is broken down prematurely.
Evaluation includes assays of fibrinolytic activity, such as euglobulin clot lysis time or specific inhibitor levels, guided by clinical suspicion.
Minimum hemostatic thresholds are factor-specific
Rare bleeding disorder data demonstrate that the minimum factor level required to prevent bleeding differs across factors.
Some factors, including fibrinogen, factor X, and factor XIII, show strong correlation between activity level and bleeding severity. Others, such as factor VII and factor XI, show weak or inconsistent correlation.
Severity cannot be defined by a universal cutoff. Each factor has its own threshold biology, and treatment targets must be individualized.
Screening tests: strengths and blind spots
PT and APTT are appropriate initial tests but have important limitations.
They may be normal in clinically significant disorders and must be followed by targeted assays when clinical suspicion persists.
Simultaneous evaluation for coagulation factor deficiencies, fibrinogen disorders, von Willebrand disease, and platelet function defects is often advantageous and reduces repeated sampling.
Testing should follow phenotype, not replace it.
Management logic follows mechanism
Management of rare bleeding disorders is shaped by heterogeneity of phenotype, limited high-quality evidence, and factor-specific thresholds.
Clinicians use replacement therapy in patients with a documented bleeding phenotype, during major trauma, in high-risk surgery, and in pregnancy and delivery.
Treatment decisions incorporate baseline factor levels, bleeding history, procedural risk, and factor-specific thresholds.
Long-term prophylaxis is generally reserved for patients with severe or recurrent spontaneous bleeding, though thresholds vary by disorder. For example, patients with factor XIII deficiency and prior intracranial hemorrhage often receive routine prophylaxis even in the absence of frequent bleeding.
Hemostasis as layered structural engineering
Initiation provides the signal.
Amplification builds the surface.
Propagation generates force.
Fibrin creates the scaffold.
Stabilization reinforces the structure.
Each layer serves a distinct function.
Each failure produces a distinct phenotype.
Mechanism explains phenotype.
Phenotype guides testing.
Testing guides therapy.
Worked example: factor XIII deficiency
In factor XIII deficiency, initiation, amplification, and propagation are intact. Thrombin is generated and fibrin forms normally. However, the fibrin scaffold is not crosslinked or protected from fibrinolysis.
The clot appears stable initially but lacks durability, leading to delayed bleeding despite normal PT and APTT. Diagnosis requires a specific factor XIII activity assay. Management involves replacement therapy, often with prophylaxis in severe cases.
Reflect & Apply
A patient has normal PT, APTT, and fibrinogen levels but develops significant bleeding 24 hours after minor surgery.
Which phase of hemostasis is most likely impaired, and which specific deficiency should be prioritized for testing given the known limitations of screening assays?
Further reading
- Palla R et al. Rare bleeding disorders: diagnosis and treatment. Blood. 2015.
- Peyvandi F et al. Rare bleeding disorders. Hematology Education. 2014.
- Hayward CPM. How I investigate for bleeding disorders. Int J Lab Hem. 2018.
Test your thinking
A short quiz on principles of hemostasis.