I posted a poll on Twitter showing a single CBC and asking for the most likely diagnosis.
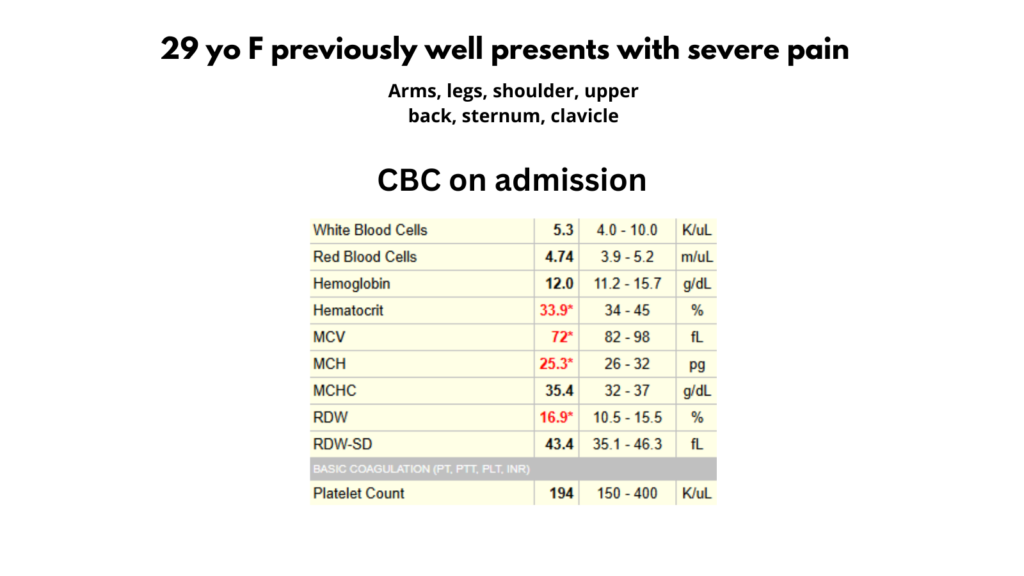
The following are the results of the poll:
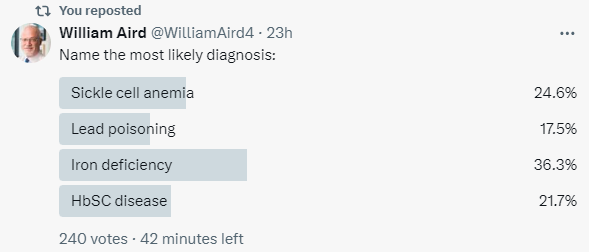
Let’s put aside the 4 possible answers/options in the poll and consider the broader differential diagnosis of microcytosis:
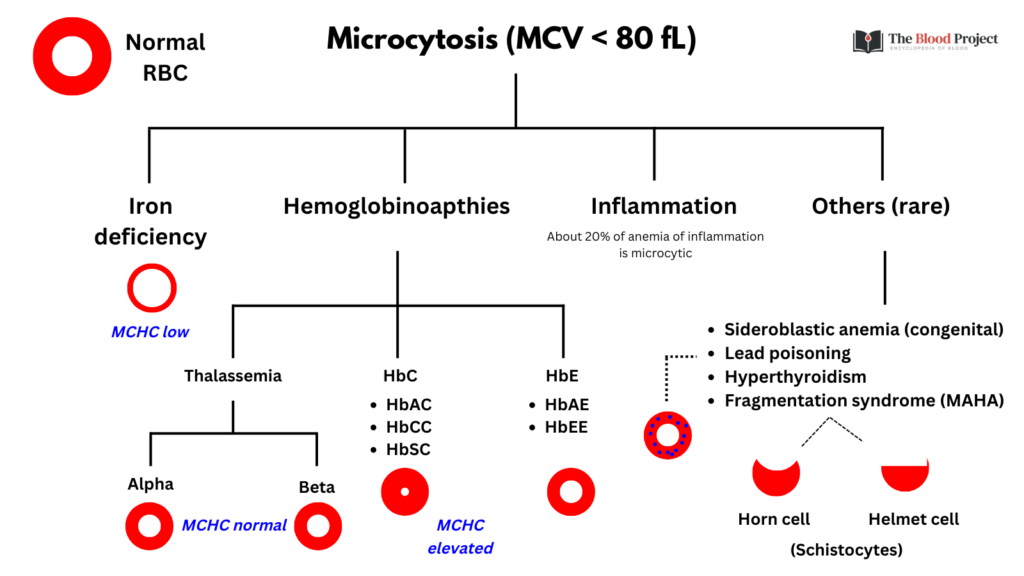
Now let’s consider the options provided in the poll:
- Sickle cell anemia (SCA):
- Term used to describe HbSS and HbSβ0-thalassemia.
- The presentation of pain is consistent with SCA.
- However, the normal Hb would be highly unusual unless the patient had been inappropriately transfused.
- SCA not associated with microcytosis.
- Lead poisoning:
- Symptoms of lead poisoning include headaches, cramps and hyperactivity.
- Almost always occurs in children, not in adults like this patient.
- May rarely cause microcytic anemia.
- Iron deficiency:
- The pain symptoms are not consistent with iron deficiency.
- The non-anemic microcytosis would be unusual, though some patients with iron deficiency manifest with an initial drop in their MCV before the Hb falls.
- The MCHC is often on the low side, and rarely as high as this patient’s 35.4 g/dL.
- HbSC disease:
- Now we’re talking!
Introduction
- Sickle cell disease (SCD) includes a group of heterogenous disorders that share the presence of the gene for hemoglobin S (HbS):1
- Homozygous condition, HbSS, is the most common type accounting for ~65% of cases in the United States.
- Compound heterozygous condition in which HbS is inherited with another abnormal hemoglobin:
- HbSC disease (the second most common type of SCD)
- Beta thalassemia:
- Hemoglobin Sβ0-thalassemia (HbSβ0-thalassemia)
- Hemoglobin Sβ+-thalassemia (HbSβ+-thalassemia)
- HbSS and HbSβ0-thalassemia are clinically very similar and therefore are commonly referred to as sickle cell anemia (SCA). While HbSC is a type of sickle cell disease, it is not considered a form of sickle cell anemia.
- Although considered a milder sickle cell disease (SCD) variant, HbSC is a sickling syndrome associated with potentially severe morbidities that warrant surveillance and intervention.2
Epidemiology
- Globally:
- The Global Burden of Disease study estimates that there are over 100,000 babies born with HbSC annually and more than 1 million affected individuals worldwide, with the majority living in low-resource settings within sub-Saharan Africa.3
- ~55000 newborns with HbSC disease are delivered yearly, with the highest hemoglobin C (HbC) gene frequency in West Africa.4
- In the United States:
- The prevalence of HbSC disease is about 1 in 7174 births.5
- 1:941 babies are born with SCD, while 1:6173 newborns has HbSC.6
- Through newborn hemoglobinopathy screening, about 600 babies with HbSC are identified in the United States each year.7
- Over 30,000 children and adults have hemoglobin SC (HbSC) disease, representing 30% of the total population with sickle cell disease.8
- One in 800 African Americans have HbSC disease.
- In the United Kingdom:
- In western Nigeria and northern Ghana:
- About 25% of the population of carry the HbC gene.12
Cause
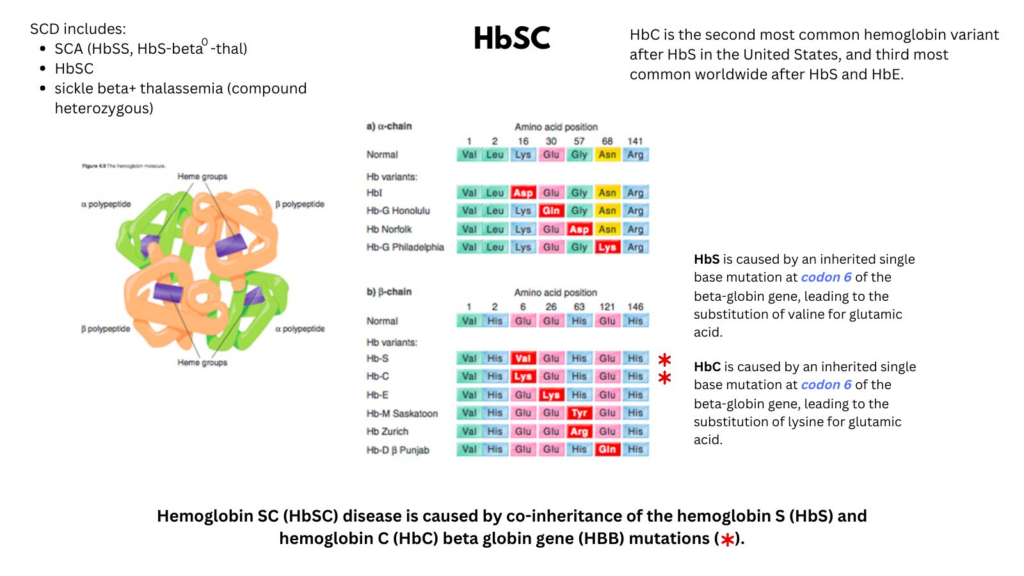
- HbSC is a double (compound) heterozygous state caused by co-inheritance of:1314
- Sickle hemoglobin (HbS; p.glu7val)
- C hemoglobin (HbC; p.glu7lys); lysine is substituted for glutamic acid (Glu6Lys) in HbC.
- Hemoglobin C (HbC) is an inherited single base mutation in codon 6 of the beta-globin gene, leading to substitution of lysine for glutamic acid. HbS is an inherited single base mutation in codon 6 of the beta-globin gene, leading to substitution of valine for glutamic acid. In HbSC one beta-globin allele is HbC, the other HbS.
- Co-inheritance of alpha-thalassemia improves red cell survival and decreases hemolysis in sickle cell disease, including HbSC.
Evolutionary Considerations
- HbSC occurs most commonly in West Africa due to the ancestral and unicentric origin of the HbC mutation that provides protection against malaria.15
- The allelic frequency of HbC is a consequence of the survival advantage against severe malaria conferred through inheritance of one HBB (beta globin gene) mutation.16
Pathophysiology
- Patients with HbSC disease have about 50% HbS and 50% HbC (with low [1–3%] HbF levels).17
- Although HbAC and HbAS are asymptomatic, their combination in HbSC is symptomatic because HbC-mediated dehydration of erythrocytes leads to polymerization of HbS and red cell sickling.1819
- HbSC disease pathogenesis is modulated by:20
- Interactions between HbS and HbC
- RBC dehydration from altered membrane transporter function, which results in increased HbS concentration, causing HbS subunit polymerization.
- Erythrocyte lifespan is twice that of HbSS (29 vs.15 days).21
Clinical Presentation
- HbSC is a sickling syndrome with an overall milder clinical phenotype than sickle cell anemia.22
- Clinical manifestations are highly varied between patients.
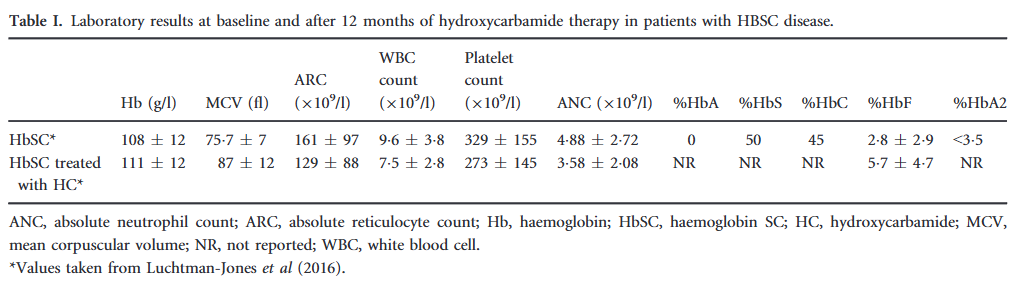
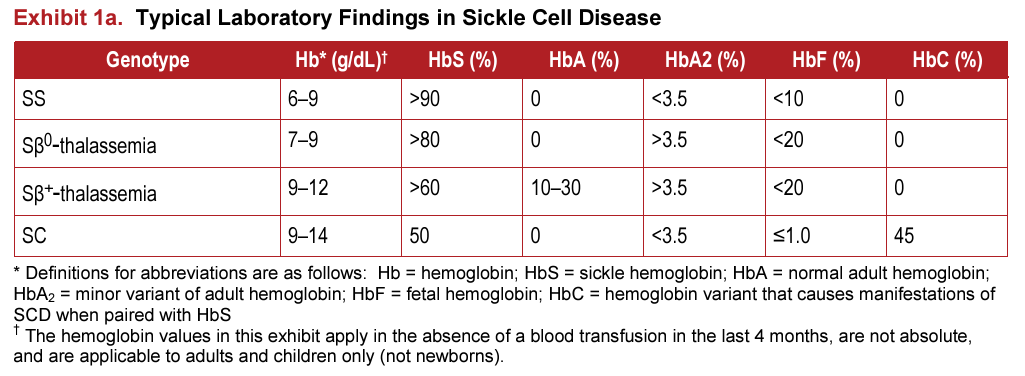
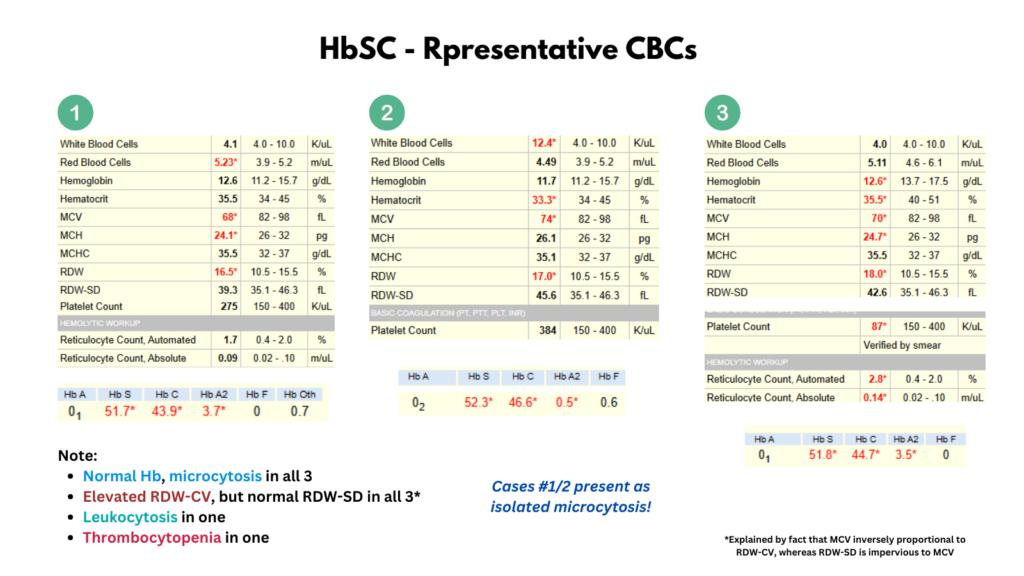
- Laboratory findings:
- Hemoglobin:
- Low mean cell volume (MCV).25
- Leukocytosis is less pronounced or absent in HbSC compared to HbSS.26
- Platelet counts may be normal, but mild to moderate thrombocytopenia occurs in patients with splenomegaly and hypersplenism.27
- Peripheral smear may show:28
- Frequent target cells from the relatively increased surface area secondary to RBC dehydration.
- Moderate microcytosis.
- Occasional microspherocytes.
- Rare distorted tri-concave or elongated erythrocytes containing Hb crystals.
- Rare irreversibly sickled cells.
- Blood viscosity is increased even compared to HbSS.29
- Erythrocyte lifespan is twice that of HbSS (29 vs.15 days).
- Acute events and chronic complications:
- Among individuals with HbSC, rates of maternal-fetal morbidity, retinopathy, avascular necrosis (AVN) of the hip, priapism, and chronic kidney disease are increased compared with historical controls.30
- Comparing HbSC with SCA:31:
- Acute vasoocclusive events such as pain and acute chest syndrome occur about half as often.
- Proliferative retinopathy, blindness, sensorineural hearing loss and thrombosis are more frequent.
- Pain crisis:32
- Hallmark of HbSC disease.
- At least 50% of people with HbSC report a painful episode requiring a hospital visit and about 5% of HbSC patients may have frequent debilitating painful events.
- Treatment strategies for painful crises in HbSC include hydration, analgesia and adequate oxygenation.
- Proliferative sickle retinopathy:33
- The most common complication of HbSC disease.
- Occurs in 30–70% of patients with HbSC compared to 3% in HbSS.
- Caused by vaso-occlusion with resultant ischemia and excessive retinal blood vessel growth.
- Incidence peaks in the third and fourth decades.
- Causes vision loss by vitreous hemorrhage and tractional retinal detachment.
- Treatment options include: diathermy, cryotherapy and transpupillary or transscleral diode laser photocoagulation.
- Annual ophthalmological examination recommended.
- Splenic complications:34
- Functional asplenia is reported in 45% of HbSC patients by age 12 years.
- Chronic splenomegaly is associated with thrombocytopenia in 35% of children and 50% of adults with HbSC and may cause recurrent abdominal pain.
- Acute splenic sequestration crisis (ASSC), precipitated by obstruction of splenic outflow with sickled erythrocytes, causes massive pooling of blood within the splenic sinusoids. ASSC occurs in 6–12% of children with HbSC.
- Osteonecrosis:35
- Aka avascular necrosis (AVN).
- Occurs in 12–24% of HbSC patients.
- Typically affects large joints, such as hips and shoulders, but can occur in other joints, including the spine.[
- AVN usually presents as focal pain.
- Reducing pain and disability are the primary management goals of AVN.
- Central nervous system complications:36
- Headache
- Ischemic stroke
- Hemorrhagic stroke
- Silent cerebral infarcts
- Priapism:37
- An unwanted erection lasting 4 h or more.
- Occurs in 20% of men with HbSC.
- Is a clinical emergency.
- Infection:
- People with HbSC and HbSβ+-thalassemia have a much lower incidence of life-threatening infection because their spleen function is normal or only minimally impaired during infancy.
- Cohort studies:
- Nelson et al, 2024
- A total of 2282 SCDIC registry participants with HbSC or SCA (HbSS and HbSß0).
- Although the HbSC participants had a lower prevalence of most SCD-related complications, they had a higher frequency of:
- Splenomegaly (n (%) = 169 (33.7) vs. 392 (22.1) for SCA)
- Retinopathy (n (%) = 116 (23.1) vs. 189 (10.6) for SCA
- The mean (standard deviation) hemoglobin level was 11.5 (1.5) g/dL among those with Hb SC versus the mean hemoglobin in the SCA cohort of 8.9 (1.5) g/dL.
- HbSC disease associated with lower markers of hemolysis with mean LDH of 314.5 (294.5) compared with 497.8 (352.5) in SCA genotypes and a total bilirubin of 1.6 (1.6) mg/dL compared with 3.3 (2.9) mg/dL.
- Among HbSC participants, there was no association be-tween hemoglobin and retinopathy or avascular necrosis.
- No association within the HbSC cohort between hemolysis biomarkers and the number of clinical complications.
- Nelson et al, 2024
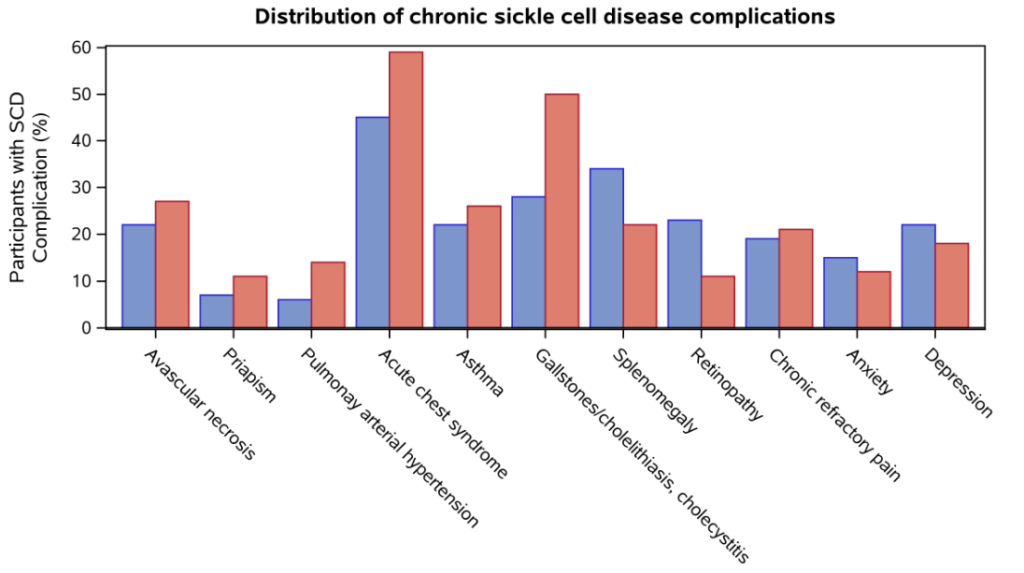
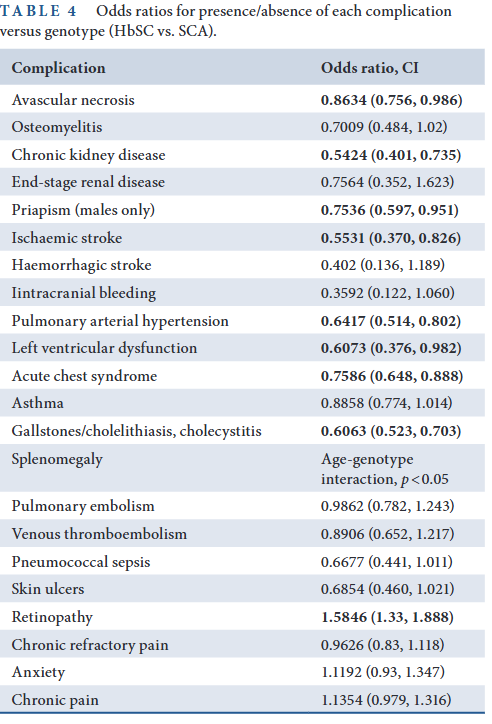
- Ghunney et al, 2023
| Characteristics | HbSC (N = 639) | HbSS (N = 639) | P value |
|---|---|---|---|
| Age, mean | 30.7 (7.8) | 31.0 (7.8) | 0.450 |
| Hb mean (SD) | 11.1 (1.3) | 8.1 (1.2) | < 0.001 |
| Acute pain events/y, n (%): | |||
| 0 events | 344 (53.8) | 289 (45.2) | < 0.001 |
| 1-2 events | 238 (37.2) | 212 (33.2) | |
| ≥3 episodes | 57 (8.9) | 138 (21.6) | |
| Pain incidence (events per 100 patient-y) | 74.6 | 123.0 | < 0.001 |
| ACS incidence (events per 100 patient-y) | 2.3 | 5.6 | 0.004 |
| AVN, n (%) | 49 (7.7) | 61 (9.5) | 0.231 |
| Cerebrovascular accident/ stroke, n (%) | 3 (0.5) | 7 (1.1) | 0.204 |
| Priapism, n (%) | 11 (4.4) | 26 (10.3) | 0.011 |
| Nephropathy, n (%) | 7 (1.1) | 54 (8.5) | < 0.001 |
| Proliferative SCD retinopathy, n (%) | 20 (3.1) | 4 (0.6) | < 0.001 |
| Chronic pain, n (%) | 110 (17.2) | 156 (24.4) | 0.002 |
Diagnosis
- Typically detected in asymptomatic newborns during routine screening.
- In absence of screening, HbSC is typically diagnosed in children, adolescents, or young adults.
- HbSC disease diagnosed by identifying significant quantities of hemoglobin C (about 50% total Hb) and hemoglobin S (about 45% of total Hb) in patient’s blood
- Can be diagnosed in laboratory by
- isoelectric focusing (IEF)
- hemoglobin electrophoresis
- high performance liquid chromatography (HPLC)
- DNA analysis
Treatment
- Lagging treatment vs. SCA
- Hydroxyurea:38
- Not as effective as in SCA.
- Small cohorts show decrease in episodes of acute chest syndrome and hospitalization for pain.
- “Moreover, disease-modifying therapies such as hydroxyurea are used less in HbSC disease compared with SCA. This is due to the lack of participants with HbSC disease in prospective drug clinical trials, which results in a paucity of efficacy and safety data in this disease subtype.”39
- Unlike HbSS, hydroxyurea therapy is not routinely recommended or used for individuals with HbSC.40
- The PIVOT trial:41
- Prospective phase 2, randomized, double-blind, placebo-controlled, non-inferiority trial in which children and adults with HbSC in Ghana were administered 12 months of hydroxyurea or placebo.
- Designed to investigate the effects of hydroxyurea treatment on sickle-related clinical and laboratory parameters in a large cohort of children and adults with HbSC disease.
- 12-month blinded treatment phase.
- After a 2-month screening period to collect pretreatment clinical and laboratory data, hydroxyurea 100-mg and 1000-mg scored tablets and matching placebo were started at 20 mg/kg of body weight as a single daily dose, with an opportunity for a 2.5 to 5.0 mg/kg dose escalation at month 2 and month 4, based on peripheral blood counts. The maximum dose of hydroxyurea was 35 mg/kg/day.
- The primary end point was hematologic dose-limiting toxicities (DLTs), including cytopenias or elevated hemoglobin levels during 12 months of blinded treatment.
- 243 patients (123 children and 120 adults).
- Almost all enrolled participants reported prior vaso-occlusive pain events similar to other descriptions of HbSC disease.
- Compared with placebo, hydroxyurea treatment was associated with:
- Conclusions: The PIVOT trial did not meet its primary end point. Hydroxyurea at 20 mg/kg in patients with HbSC was associated with more hematologic DLTs than placebo, but most were mild and transient. Hydroxyurea was associated with less vaso-occlusive pain and fewer sickle-related events in both children and adults; a new trial will need to be done to establish the efficacy of this approach.
- Phlebotomy in those with high hemoglobin values and increased in blood viscosity.44
Prognosis
- Median survival in resource-rich countries has risen to 80 years.45
- Survival of patients with HbSC is superior to SCA.46
- The PIVOT trial:47
Guideline recommendations
- 2014 National Heart, Lung, and Blood Institute (NHLBI)-sponsored expert panel evidence-based report on management of sickle cell disease

Health Maintenance for People With Sickle Cell Disease
| SCA | HbSC | |
|---|---|---|
| Oral penicillin prophylaxis | Administer oral penicillin prophylaxis (125 mg for age <3 years and 250 mg for age ≥3 years) twice daily until age 5 in all children with HbSS | Consider withholding penicillin prophylaxis from children with HbSC disease and HbSβ+-thalassemia unless they have had a splenectomy |
| Vaccination against Strep pneumoniae | Assure that people of all ages with SCD have been vaccinated against Streptococcus pneumoniae | Same as with SCA |
| Proteinuria screen | Screen all individuals with SCD, beginning by age 10, for proteinuria. | Same as with SCA |
| EKG | Routine ECG screening is not recommended in children and adults with SCD. | Same as with SCA |
| Ophthalmology screen | In people with SCD, refer to an ophthalmologist for a dilated eye examination to evaluate for retinopathy beginning at age 10; For people having a normal dilated retinal examination, re-screen at 1–2 year intervals. | Same as with SCA |
| Transcranial Doppler (TCD) imaging | In children with SCA, screen annually with TCD according to methods employed in the STOP studies, beginning at age 2 and continuing until at least age 16. | In children with genotypes other than SCA (e.g., HbSβ+-thalassemia or HbSC), do not perform screening with TCD. |
| Pulmonary function tests | Do not screen asymptomatic children and adults with pulmonary function tests. | Same as with SCA |
Managing Acute Complications of Sickle Cell Disease
| SCA | HbSC | |
|---|---|---|
| Vaso-Occlusive Crisis | ||
| Pain control | In adults and children with SCD and a VOC associated with severe pain, rapidly initiate treatment with parenteral opioids. | Same as with SCA |
| Anemia | Use simple transfusion in people with SCD and acute anemia whose symptoms are due to anemia. | Same as with SCA |
| Acute chest syndrome | Treat people with SCD who have ACS with an intravenous cephalosporin, an oral macrolide antibiotic, supplemental oxygen (to maintain oxygen saturation of greater than 95 percent), and close monitoring for bronchospasm, acute anemia, and hypoxemia. In people with SCA, give simple blood transfusion (10 mL/kg red blood cells) to improve oxygen carrying capacity to people with symptomatic ACS whose hemoglobin concentration is >1.0 g/dL below baseline. If baseline hemoglobin is 9 g/dL or higher, simple blood transfusion may not be required. n all persons with SCD, perform urgent exchange transfusion—with consultation from hematology, critical care, and/or apheresis specialists—when there is rapid progression of ACS | In people with HbSC disease or HbSβ+-thalassemia with ACS, decisions about transfusion should be made in consultation with an SCD expert. |
Hydroxyurea


For quiz, click here.