The diagnosis of thalassemias relies on a stepwise approach, beginning with clinical suspicion and routine blood tests, and moving toward confirmatory studies such as hemoglobin electrophoresis or genetic testing. Because some findings overlap with other causes of microcytosis, particularly iron deficiency, careful evaluation is required to distinguish between them and to determine the specific type and severity of thalassemia.
Overview
Clinical Suspicion
- Family history of anemia, microcytosis, or thalassemia
- Geographic/ethnic background – Mediterranean, Middle Eastern, South Asian, African, Southeast Asian
- Presentation:
- Trait → asymptomatic, incidental finding of microcytosis, sometimes have mild anemia
- Non transfusion dependent thalassemia (NTDT) → mild–moderate anemia, splenomegaly
- Transfusion dependent thalassemia (TDT) → severe anemia in infancy
- Typical presentation
- Age of onset: Usually between 6–24 months.
- Key features:
- Severe microcytic anemia
- Mild jaundice
- Hepatosplenomegaly
- Clinical manifestations:
- Failure to thrive
- Progressive pallor
- Feeding difficulties and irritability
- Recurrent fevers (hypermetabolic state or intercurrent infection).
- Abdominal distension due to splenomegaly and hepatomegaly.
- Clinical picture of untreated or poorly transfused thalassemia:
- General features:
- Growth retardation
- Pallor
- Jaundice
- Poor musculature
- Genu valgum (knock knees)
- Organ-related findings:
- Hepatosplenomegaly
- Leg ulcers
- Masses due to extramedullary hematopoiesis
- Skeletal changes (bone marrow expansion):
- Long bones: deformities in legs
- Craniofacial abnormalities (“thalassemic facies”)
- General features:
- Typical presentation
Initial Laboratory Clues
- Complete blood count:
- Microcytosis (low MCV) disproportionate to hemoglobin level
- Elevated RBC count (especially in trait)
- Peripheral smear:
- Microcytosis
- Hypochromia
- Target cells
- Reticulocytes: elevated in NTDT/TDT, often blunted relative to anemia severity
Rule Out Iron Deficiency
- Iron studies: Ferritin, serum iron, TIBC, transferrin saturation
- Normal or ↑ ferritin with microcytosis → favors thalassemia over iron deficiency
Differentiating Iron Deficiency from Thalassemia Trait
While low MCV and MCH are characteristic of thalassemic red blood cells, these indices alone cannot distinguish between thalassaemia trait and iron deficiency and the hematological parameters in these two conditions may closely resemble each other, leading to confusion between α-thalassaemia trait and iron-deficiency anaemia. In such cases, assessing iron status (e.g., serum iron, transferrin saturation, or red blood cell zinc protoporphyrin levels) can be helpful in making an accurate diagnosis although it may not be sufficient in those where the two conditions co-exist. alpha CPG
Many formulas have been proposed to discriminate between iron deficiency anemia (IDA) and β-thalassemia trait (BTT). While widely cited, most have not been validated in α-thalassemia. Clinically, these formulas are used as bedside tools to help estimate the pretest probability of one versus the other, rather than to make a definitive diagnosis.
Rather than simply plugging numbers into a formula, it pays to understand the pathophysiological underpinnings. Almost all of these indices are built from some combination of MCV (mean corpuscular volume) and RBC count:
- In β-thalassemia trait, the red cells are microcytic. However, the bone marrow adapts by producing a larger number of red blood cells, so that total hemoglobin/hematocrit is relatively preserved despite the smaller cell size. Thus, the RBC count is normal or high relative to the degree of microcytosis.
- In iron deficiency anemia, the red cells are also microcytic, and the marrow wants to increase red cell production. But because iron is an essential substrate for hemoglobin synthesis, the marrow cannot expand production. As a result, the RBC count tends to be low or low-normal, out of proportion to the degree of microcytosis.
The Mentzer Index is the most famous of these formulas. It is defined as MCV (fL)/RBC count (millions/μL)
- < 13 → favors β-thalassemia trait
- > 13 → favors iron deficiency anemia
This works because in thalassemia, the denominator (RBC count) is relatively high, driving the ratio down, whereas in iron deficiency, the denominator is low, driving the ratio up.
Other indices (e.g., Shine & Lal, Srivastava, Green & King, RDWI) combine MCV, MCH, RDW, and Hb values in similar ways, but all exploit the same underlying principle: microcytosis with preserved/increased RBC count suggests thalassemia trait; microcytosis with reduced RBC count suggests iron deficiency.
Clinicopathological features indicating the need for haemoglobinopathy investigations (BJH Clinical Practice Guidelines; exclui9dng sickle cell disease and high affinity hemoglobin)efn_note]PMID: 37271570[/efn_note]
| Indication | Suspected globin chain disorder |
|---|---|
| Hydropic fetus | Hemoglobin Bart’s hydrops fetalis |
| Neonate or infant with anemia and either Hb F only or unexpectedly low percentage of Hb A | β thalassemia major/TDT |
| Unexplained anemia and splenomegaly | β thalassemia major/TDT or intermedia/NTDT, hemoglobin H disease, unstable hemoglobin |
| Suspected thalassemia or unexplained microcytosis | Thalassemia or thalassemic haemoglobinopathy including hemoglobin H disease, hemoglobin E and hemoglobin Lepore |
| Unexplained hemolysis | Hemoglobin H disease, unstable hemoglobin |
| Unexplained target cells | Thalassemia, variant hemoglobin |
| Unexplained irregularly contracted cells | Variant hemoglobin, particularly hemoglobin C, hemoglobin E or an unstable hemoglobin |
Screening for thalassemia1
“Methods used are red cell indices in conjunction with the measurement of Hb A2 and Hb F percentages. Routine
measurement of indices includes measurements of MCH and mean cell volume (MCV); it is recommended that
MCH is used to screen for thalassaemia as this parameter is more stable than MCV. These measurements are
reported for all commonly used automated instruments. Hb A2 is quantified by HPLC or CE. While the Hb F may
be raised, it is the Hb A2 percentage that is most significant for the diagnosis of β thalassemia trait. Hb F percentage is particularly important in the diagnosis of δβ thalassemia… A national recommended value of Hb A2 of 3.5% or
above has been set as the action point in the diagnosis of carriers of β thalassemia in the antenatal screening program… Hb A2 values >4.0% with normal indices may indicate β thalassaemia trait with or without co-existing α thalassaemia, or with co-existing conditions such as vitamin B12 or folate deficiency”Confirmatory Testing
- Hemoglobin electrophoresis / HPLC:
- β-thalassemia trait: ↑ HbA₂ (>3.5%), sometimes ↑ HbF
- β-thalassemia major/intermedia: ↑↑ HbF, little/no HbA
- α-thalassemia trait: usually normal electrophoresis → diagnosis of exclusion unless genetic testing done
- Genetic testing:
- α-thalassemia: detects gene deletions
- β-thalassemia: identifies point mutations
- Increasingly used for definitive diagnosis, family counseling, and prenatal diagnosis
Additional/Supportive Studies
- Bone marrow (rarely needed): Erythroid hyperplasia, ineffective erythropoiesis
- Imaging: Splenomegaly, extramedullary hematopoiesis in severe cases
- Iron assessment: Ferritin, MRI for liver/cardiac iron (especially in NTDT/TDT)
Diagnosis of Specific Types of Thalassemia
Diagnosis of alpha-thalassemia carrier and trait
- -α/αα carriers may have normal red cell indices or only slightly reduced MCV and MCH levels.
- Those with genotypes -α/-α and –/αα typically exhibit reduced MCV and MCH values.
Diagnosis of beta-thalassemia trait
- Red cell indices:
- Low mean corpuscular hemoglobin (MCH)
- Low mean corpuscular volume (MCV)
- Hemoglobin: low-normal or slightly subnormal
- Peripheral blood film:
- Mild erythrocyte morphological changes (less severe than in affected individuals)
- Nucleated red blood cells typically absent
- Elevated HbA₂ levels
Diagnosis of beta-thalassemia intermedia
- Beta-thalassemia intermedia suspected in individuals who present with similar but milder symptoms at a later age compared to beta-thalassemia major, or with emerging complications
- Hematologic features of beta-thalassemia intermedia include:
- Hb level 7-10 g/dL (70-100 g/L)
- MCV between 50 and 80 fL
- MCH between 16 and 24 pg
- In beta-thalassemia intermedia hemoglobin electrophoresis or high-performance liquid chromatography (HPLC) shows
- 10%-30% HbA
- 2%-5% HbA2
- 70%-90% HbF2%-5% HbA2
- 70%-90% HbF
Diagnosis of beta-thalassemia major
- Beta-thalassemia major suspected in children < 2 years old presenting with mild jaundice, hepatosplenomegaly, and severe microcytic anemia
- Hematologic features of beta-thalassemia major include
- Hemoglobin (Hb) level < 7 g/dL (70 g/L)
- Mean corpuscular volume (MCV) between 50 and 70 femtoliters (fL)
- Mean corpuscular hemoglobin (MCH) between 12 and 20 pg
- Peripheral smear:
- Microcytosis
- Hypochromia
- Anisocytosis
- Poikilocytosis
- Nucleated red blood cells (NRBCs, erythroblasts)
- In beta-thalassemia major, hemoglobin electrophoresis or HPLC shows:
- Absent or very low HbA
- 2%-5% HbA2
- 92%-95% HbF
Thalassemia Variants
- Abnormal red cell morphology is common across β-thalassemia, α-thalassemia, and compound states.
- Hb E/β-thalassemia: Shares similar features.
- Hb E detection: Can be readily identified with dichlorophenolindophenol dye.
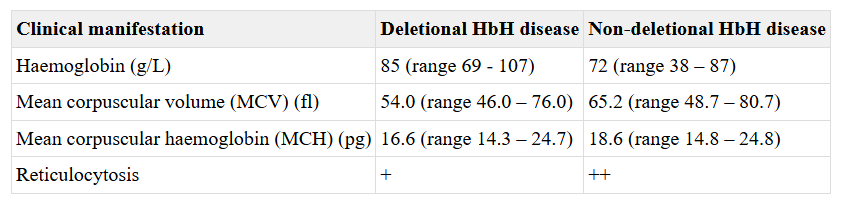
Diagnosis of HbH disease
- Suspect in HbH disease in patient with:
- Supravital staining of peripheral blood will show presence of red blood cell inclusion bodies (HbH precipitate formed from beta4 tetramers) in 5%-80% of erythrocytes
- Qualitative and quantitative hemoglobin analysis may show variable levels of HbH (0.8%-40% of total Hb) and reduction in HbA levels (60%-90%)
- Genetic testing will usually show deletion/inactivation of 3 alpha-globin genes
Diagnosis of Barts
- Usually established in the fetus
- Should be suspected in at-risk pregnancy at 13-14 weeks gestation with presence of increased (by ultrasound):
- Nuchal thickness
- Placental thickness
- Middle cerebral artery velocity
- Cardiothoracic ratio
- Ultrasound findings consistence with development of hydrops that usually becomes evident at 22-28 weeks gestation include:
- Generalized edema
- Ascites
- Pleural effusions
- Pericardial effusions
- Other prenatal testing results that supportive of diagnosis:
- Fetal blood sampling for complete blood count which may show severe hypochromic anemia
- Qualitative and quantitative hemoglobin analysis will show Hb Bart level 85%-90% in fetal blood and 100% in newborns
- Genetic testing from chorionic villi or cultured amniotic fluid cells will most commonly show deletion of all 4 alpha-globin genes
Testing Details
Complete Blood Count
- Hemoglobin (Hb):
- Normal or mildly reduced in trait
- Moderate in NTDT
- Severely reduced in TDT
- Mean corpuscular volume (MCV): Markedly ↓ (microcytosis), often out of proportion to Hb level
- Mean corpuscular hemoglobin (MCH): ↓
- Red cell count (RBC): Relatively ↑ (especially in trait) despite anemia
- RDW: Normal or mildly ↑ (contrast with iron deficiency, where RDW is often high)
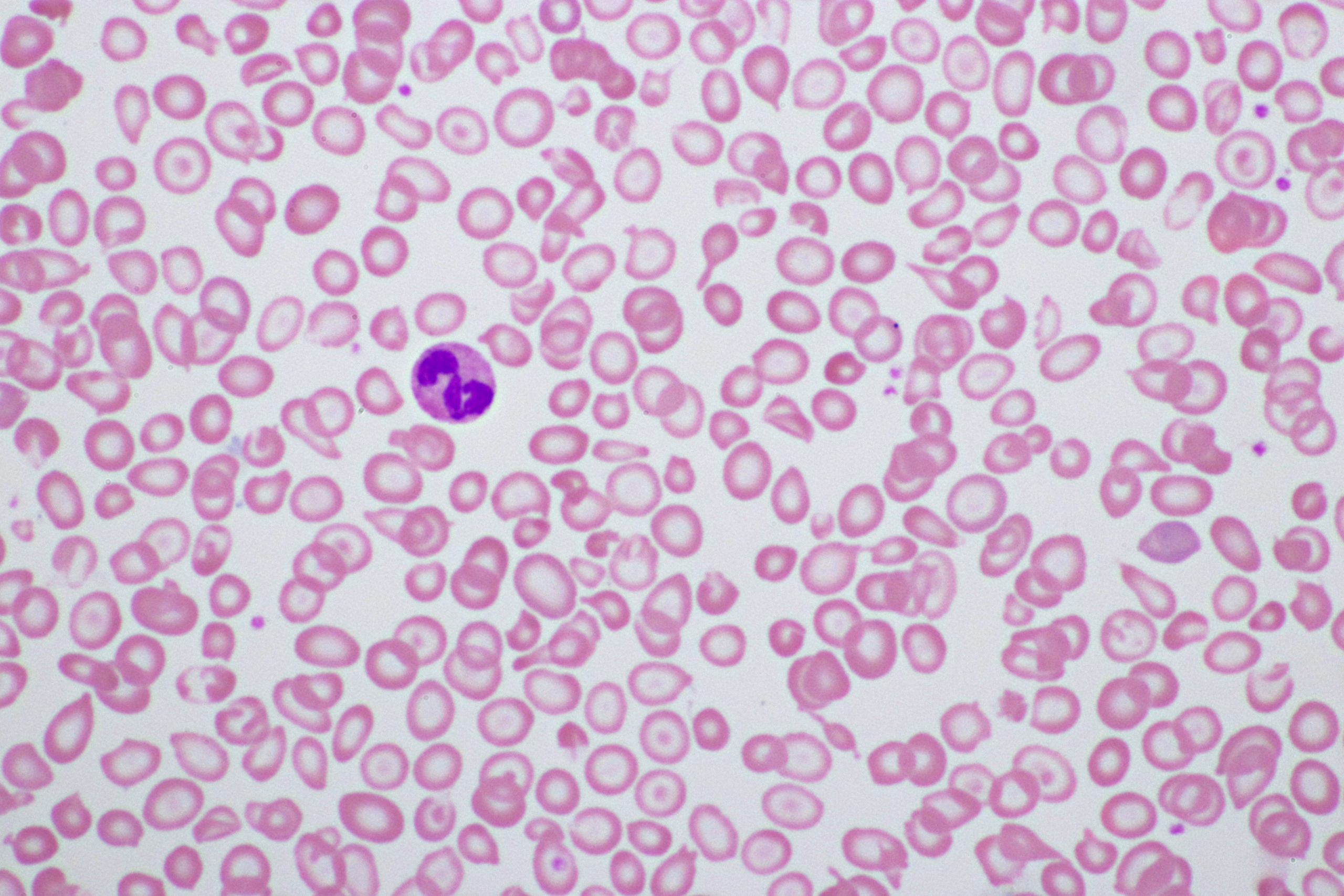
Peripheral Blood Smear
- Size:
- Microcytosis – uniformly small red cells
- Anisocytosis – especially in more severe forms (NTDT, TDT)
- Staining:
- Hypochromia – pale cells with large central pallor
- Polychromasia – variable, depending on reticulocyte output
- Poikilocytes – target cells, especially in β-thalassemia
- Inclusions:
- Basophilic stippling – especially in β-thalassemia
- Nucleated RBCs – in more severe forms (NTDT, TDT) reflecting marrow stress
- Marked changes with severity:
- Trait: subtle microcytosis, target cells
- NTDT: microcytosis, target cells, anisopoikilocytosis, occasional nucleated RBCs
- TDT: severe microcytosis, target cells, nucleated RBCs, marked anisopoikilocytosis
Iron studies
- Evaluation of microcytic anemia:
- During evaluation of microcytic anemia, serum ferritin level commonly used to rule out iron deficiency anemia; in patients presenting with microcytic anemia; serum ferritin ≤ 12 ng/mL may indicate iron deficiency anemia.
- Other iron tests that can help support diagnosis of iron deficiency anemia include evaluation of serum iron and TSAT.
- In patients with findings consistent with iron deficiency anemia, if no improvement in anemia is observed after 3 months of iron supplementation with improvement in iron deficiency, consider hemoglobin analysis
- Evaluation of iron overload:
- Serum ferritin:
- Serum ferritin usually correlates with body iron stores, but may not be a reliable marker of iron overload as a single value because increased value may also correlate with presence of inflammatory disorders and liver disease.
- Serial measurements are more reliable for assessing iron overload and efficacy of chelation.
- Serum ferritin ≥ 800 ng/mL reported to be associated with increased risk of serious iron-related complications and may warrant iron chelation therapy in patients age ≥ 10 years (patients with non-transfusion-dependent beta-thalassemia.
- Transferrin saturation (TSAT):
- Measures available iron.
- 20%-50% is considered normal.
- > 50% indicates iron overload.
- Values > 70%-75% usually indicates presence of non-transferrin-bound iron (NTBI).
- Serum ferritin:
Qualitative and Quantitative Analysis of Hemoglobin
- Methods based on physical separation of hemoglobin (Hb) to examine properties of Hb subunits:
- Specific standards such as HbA, HbS, HbC, HbF, and HbA2 are evaluated alongside patient samples as controls
- Can identify other hemoglobin variants such as Hb S, C, E, OArab, and Lepore that may co-exist with beta-thalassemia
- Techniques include:
- Hb electrophoresis:
- Capillary electrophoresis
- Methods:
- The CE system is based on capillary electrophoresis in free solution from cathode to anode. Hemoglobin components are separated in silica capillaries by their electroosmotic flow and at a high voltage (9,800 V) in electrophoretic mobility in an alkaline buffer. The photometry at an absorbance wavelength 415 nm is used to directly detect Hb fractions.2
- Core Capabilities
- Quantifies Hb A, A2, F, and S, and detects/provisionally identifies many variant hemoglobins
- Provides accurate Hb A2 quantification, suitable for diagnosing β-thalassemia trait
- Separates Hb E from Hb A2, allowing accurate A2 measurement in the presence of Hb E (but not Hb C)
- Typically separates:
- Hb A, A2, E, F, S, C, D-Punjab, and G-Philadelphia
- Note: Overlap between Hb C and Hb A2 can occur
- Practical Advantages:
- Automated CE systems are increasingly used as the initial diagnostic method in high-throughput labs
- Can handle a larger workload than HPLC
- Does not separate glycosylated hemoglobins
- Considered an advantage in avoiding interpretive complications related to post-translational modifications
- Automated CE systems are increasingly used as the initial diagnostic method in high-throughput labs
- Interpretive Considerations
- Optical density > 0.07 and presence of Hb A + A2 or Hb F + A2 is required for:
- Zone identification
- Provisional identification of variants
- Results may differ from HPLC
- Due to HPLC’s ability to separate glycosylated/post-translationally modified hemoglobins, percentages of variants and normal Hb may not match across platforms
- Optical density > 0.07 and presence of Hb A + A2 or Hb F + A2 is required for:
- Quality Control & Follow-Up:
- Careful review of chromatograms is essential
- Controls must be run with every batch
- Variant identification by CE is provisional
- Confirmatory testing should include:
- HPLC
- Alkaline and acid pH electrophoresis
- Sickle solubility test (if Hb S is suspected)
- Molecular techniques (if needed for definitive diagnosis)
- Confirmatory testing should include:
- Methods:
- Cellulose acetate electrophoresis:
- Utility and Applications
- Simple, rapid, and reliable method for hemoglobin separation
- Still useful in resource-limited settings and as a secondary technique (e.g., after HPLC)
- Enables provisional identification of common hemoglobins:
- Hb A, F, S/G/D, C/E/O-Arab, Hb H
- Also identifies less common variants
- Declining Use:
- Use is in steady decline in high-income countries due to:
- Limited throughput
- Inability to keep up with high-volume workloads compared to automated systems like HPLC and CE
- Use is in steady decline in high-income countries due to:
- Interpretive Considerations
- A single band in the S or C position could reflect:
- A compound heterozygous state (e.g., S/D, C/E, etc.)
- Hb variant + β⁰-thalassemia, especially if microcytosis is present
- Hb S can be quantified by scanning densitometry after staining
- Hb A2 quantification is not recommended using this method:
- Lacks precision necessary for diagnosing β-thalassemia trait
- A single band in the S or C position could reflect:
- Capillary electrophoresis
- High-performance liquid chromatography (HPLC) or DE-52 microchromatography
- Methods:
- The HPLC system is cation exchange and use two dual piston pumps to set gradient sodium phosphate buffers of increasing ionic strength to pass through a column spherical cation exchange resin during a 6.5 minutes. Hemolysate samples are determined by spectrophotometer that read double wavelengths at 415 and 690 nm. The resulting chromatograms are separated in retention time (RT).3
- Advantages and common uses:
- Quantifies Hb A, A2, F, and S, and detects/provisionally identifies many variant hemoglobins.
- Especially effective at accurately measuring Hb A2, making it suitable for diagnosing β-thalassemia trait.
- Commonly adopted in high-throughput labs as the initial diagnostic tool.
- Typically separates hemoglobins such as: A, A2, F, S, C, D-Punjab, and G-Philadelphia
- Limitations and interferences:
- Co-elution issues:
- Hb E and Hb Lepore often co-elute with Hb A2.
- Other variants may co-elute with Hb A, S, or F, requiring alternative techniques for clarification.
- Separates glycosylated and derivative forms of hemoglobin:
- May confound interpretation, especially in cases involving Hb S.
- For example, glycosylated Hb S can appear in the A0 window, leading to misinterpretation as Hb A.
- In patients with sickle cell anemia or sickle/β-thalassemia, reporting a low % in the A0 window is discouraged due to potential clinical confusion.
- Co-elution issues:
- Quantification Caveats
- Quantification Caveats
- Some HPLC systems:
- Do not accurately quantify Hb H
- Underestimate Hb F
- May not detect adducts of Hb F
- As a result:
- The total of all hemoglobins (normal + variant) may not equal 100%
- This discrepancy is method-dependent
- Quality Control and Confirmation
- Controls must be run with every batch.
- Identification of variants is provisional and should be followed by second-line methods:
- Capillary electrophoresis (CE)
- Alkaline and acid pH electrophoresis
- Sickle solubility test (if Hb S is suspected)
- Molecular methods (e.g., DNA or protein sequencing) for definitive confirmation
- Methods:
- Hb electrophoresis:
- Detection of hemoglobin H bodies (Hb H bodies):4
- Definition & Detection
- Hb H bodies are intracellular precipitates of β₄ tetramers.
- Detected in red blood cells after supravital staining with:
- Brilliant cresyl blue, or
- New methylene blue
- Appear as golf-ball-like inclusions in affected red cells
- Clinical Context
- Seen in conditions with excess β-globin chains, typically due to:
- α-globin deficiency
- Disorders where Hb H bodies may be detected:
- Hb H disease
- Carriers of α-thalassemia
- ATRX syndrome (α-thalassemia with mental retardation)
- Acquired Hb H disease
- Declining Use for Genotyping
- Historically used to distinguish between:
- Homozygous α⁺-thalassemia (−α/−α)
- Heterozygous α⁰-thalassemia (−−/αα)
- These conditions are hematologically similar, but only homozygous α⁰ can lead to Hb Bart’s hydrops fetalis.
- Hb H body detection is unreliable and labor-intensive for this purpose and has been superseded by DNA analysis.
- As a result, many labs have discontinued this manual technique.
- Historically used to distinguish between:
- Current Clinical Utility
- Still useful in:
- Confirming Hb H disease (~5% of red cells show inclusions)
- Supporting diagnosis of:
- ATRX syndrome (occasional Hb H bodies)
- Acquired Hb H disease (often >5% of cells affected)
- Particularly helpful when patients have unexplained microcytosis or hypochromia with characteristic clinical findings
- Seen in conditions with excess β-globin chains, typically due to:
- Definition & Detection
- Typical findings:
- HbF levels:
- Beta thalassemia:
- Up to 100% of total Hb in beta-thalassemia major and 10%-50% (but can be up to 100%) in beta-thalassemia intermedia
- May also depend on genotype:
- 92%-95% of total Hb in beta0 thalassemia homozygotes
- 70%-90% of total Hb in beta+ thalassemia homozygotes or beta+/beta0 compound heterozygotes
- Beta thalassemia:
- Hb A levels:
- Beta thalassemia:
- Absent in beta0 thalassemia homozygotes; diagnosis of 0 thalassemia homozygotes can be made at birth due to complete absence of HbA
- 10%-30% of total Hb in beta+ thalassemia homozygotes or beta+/beta0 compound heterozygotes; due to residual levels of HbA that overlaps with reference range for neonates, identification cannot be made at birth based on HbA levels
- Beta thalassemia:
- Hb A2 levels:
- Beta thalassemia:
- typically 2%-5% of total Hb
- HbA2 levels may be lower in beta-thalassemia major (< 3.5% of total Hb) compared to beta-thalassemia intermedia (≥ 3.5%)
- Beta thalassemia:
- HbF levels:

Hb electrophoresis. Source
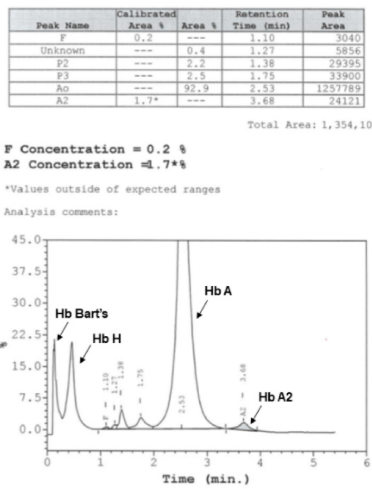
HPLC. Source
Genetic testing
- Indications for testing:
- In patients with suspected beta-thalassemia major or intermedia:
- confirmation of beta-thalassemia diagnosis
- predicting disease severity based on specific mutations
- preconception genetic counseling for at-risk families
- As part of carrier screening including family members, gamete donors, and members of at-risk ethnic group
- In patients with suspected beta-thalassemia major or intermedia:
- Methods include:
- Targeted polymerase chain reaction (PCR)-based methods:
- Mutation-specific detection, cannot detect unknown or rare gene variations
- Consider using this method in geographic regions where number of known pathogenic beta-globin gene mutations are limited.
- Gene sequence analysis (e.g., Sanger sequencing of the β-globin gene):
- consider using this method if rare mutation is suspected or if targeted analysis did not identify known mutation
- testing methods include direct sequencing or array analysis
- Next-Generation Sequencing (NGS):5
- Increasingly used in commercial diagnostic kits.
- Advantages over Sanger:
- Does not require prior knowledge of target sequence.
- Eliminates need for mutation-specific primers.
- Process:
- Multiplex PCR → DNA fragment (amplicon) library.
- Ligation of amplicons to adapters with unique index sequences.
- High-throughput sequencing.
- Detects:
- Single nucleotide variants (SNVs).
- Insertions/deletions (indels).
- Copy number variations (CNVs) in globin genes.
- Large upstream/downstream deletions or duplications (e.g., in εγδβ-thalassemia)
- Additional information on laboratory testing, including molecular testing methods for thalassemias can be found
- Targeted polymerase chain reaction (PCR)-based methods:
Clinical practice guidelines6
- A diagnosis of thalassaemia should be considered in all those who have hypochromic microcytic anaemia (Grade C, Class I).
- In the diagnostic work-up for hypochromic microcytosis, iron deficiency anaemia should always be excluded (Grade C, Class I).
- Molecular analysis is not required to confirm the diagnosis of a β-thalassaemia carrier, but it is necessary to confirm the α-thalassaemia carrier status(Grade C, Class I).
- An α-globin gene triplication or quadruplication should be taken into consideration in heterozygous β-thalassaemia subjects with a β-thalassaemia intermedia phenotype (Grade C, Class I).
- Haematological parameters, including red cell indices and morphology, followed by separation and measurement of haemoglobin fractions, are the basis for the identification of β thalassaemia carriers (Grade C, Class I).
- Since the prevalent pathogenic variants of the β-globin gene are limited in each at-risk population, a polymerase chain reaction (PCR) method designed to detect the common specific mutation simultaneously should be used initially (Grade C, Class IIa).
- β-globin gene sequence analysis may be considered first if the affected individual is not of an ancestry at high risk or if targeted analysis reveals only one or no pathogenic variant (Grade C, Class IIa).
- Considerations of phenotype should not only be based on genotype but should take clinical presentation and disease severity as observed over a duration of time (Grade C, Class IIb).
- Patients who receive 6 or more red blood cell units over 6 months with ≤6-week transfusion free period or receiving frequent transfusions for >1 year can be classified as transfusion dependent β-thalassaemia for purposes of management approach or clinical trial eligibility (Grade C, Class IIb).
British Society for Haematology Guideline: Significant haemoglobinopathies: A guideline for screening and
diagnosis:7
- Patients who present for diagnosis rather than for screening, who have microcytosis, should be evaluated clinically with further testing for iron deficiency, anemia of chronic disease and thalassemia being carried out in the light of the red cell indices and the clinical findings. (1B)
Alpha CPG
- Accurate diagnosis of α-thalassaemia syndromes requires application of a range of diagnostic techniques, including complete blood count (CBC) with reticulocyte count, haemolytic panel, peripheral blood smears, automatic haemoglobin analyzers, and different modalities of molecular analysis.