Stopping hemolysis at the point of expression; blocking the effector rather than the source
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
Why this spoke matters
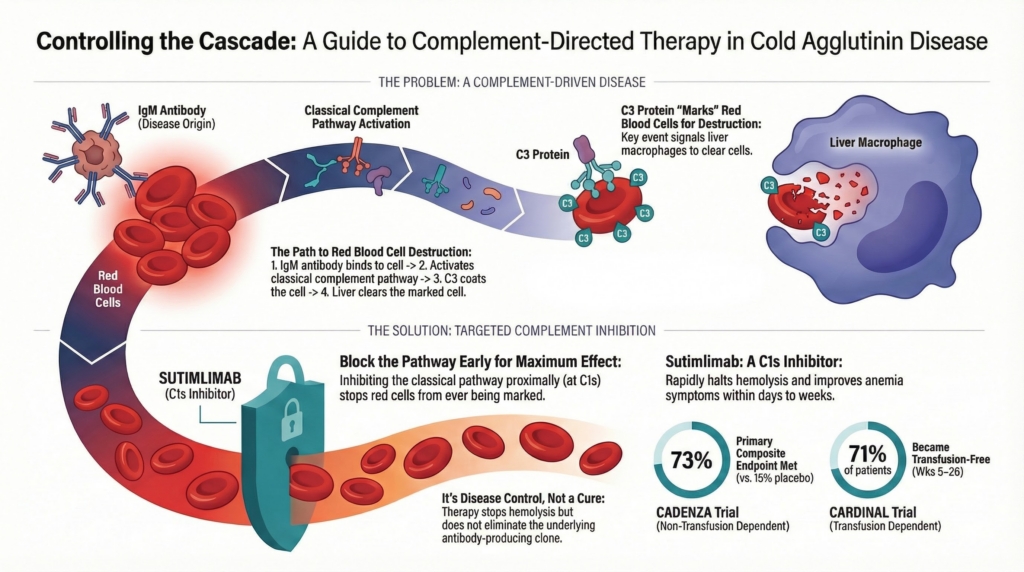
Cold agglutinin disease is not explained by antibody quantity alone; disease expression depends on downstream classical-pathway complement activation after antibody binding. It is best understood as a complement-driven hemolytic disorder in which antibody binding initiates the process, but complement activation determines whether and how disease is expressed. In primary CAD, IgM binding activates the classical pathway, leading predominantly to C3 deposition on red cells, with more limited terminal complement activation and MAC formation, particularly during severe disease or acute exacerbations.1 Classical pathway–mediated C3 deposition is therefore the dominant driver of red-cell destruction and clinical hemolysis. Disease severity is not reliably predicted by IgM titer alone; it reflects the extent of downstream complement activation together with antibody characteristics such as thermal amplitude.2
Complement-directed therapy directly targets the effector phase of hemolysis in CAD.3
This framing sets up a recurring tension in CAD management: whether the immediate goal is to stop hemolysis now through complement inhibition, or to pursue longer-term disease modification by targeting pathogenic IgM production with clone-directed therapy.4
That distinction also anticipates a central theme in CAD: hemolysis and circulatory symptoms arise from different mechanisms and may respond differently to therapy. Anemia reflects complement-mediated red-cell clearance, whereas cold-induced circulatory symptoms often reflect IgM-mediated agglutination and impaired microvascular flow.5
The current spoke explains what complement inhibition does, why it works, and where its limits lie.
The biological target
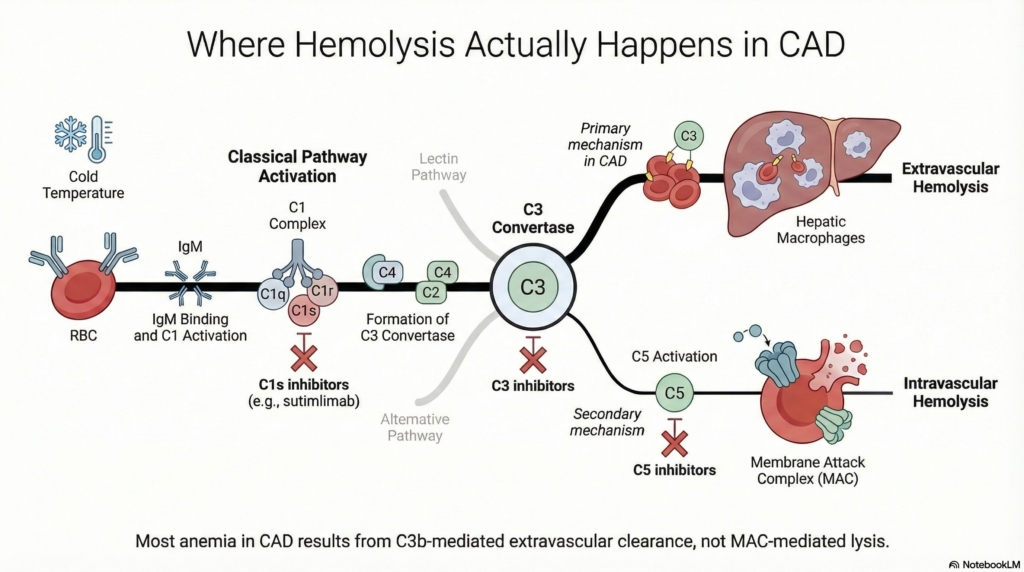
In primary cold agglutinin disease, the pathogenic IgM antibody binds red cells at lower temperatures and initiates two related downstream processes. Classical complement activation determines the hemolytic phenotype, including C3 deposition, hepatic red-cell clearance, anemia, and hemolysis markers. IgM-mediated red-cell agglutination in cooler vascular beds contributes to cold-induced circulatory symptoms.6
Key features of complement involvement include:7
- activation through the classical pathway
- C3b-mediated opsonization of red cells as the dominant effector step in chronic hemolysis
- hemolysis that is predominantly extravascular, with hepatic clearance of C3b/iC3b-opsonized red cells
- a limited contribution from terminal complement–mediated intravascular hemolysis in steady-state disease, although this may increase during severe exacerbations
Complement-directed therapy intervenes downstream of antibody binding but upstream of red-cell destruction, targeting the point at which disease is actually expressed.8
Available complement-directed agents in CAD
Understanding the biological target makes the therapeutic logic clear. Because disease expression in CAD is driven by classical pathway activation and C3-mediated opsonization, effective complement-directed therapy must interrupt the cascade upstream of C3 deposition, before red cells are marked for clearance.9
This requirement narrows the field of effective agents and explains why proximal classical pathway inhibition has proven clinically meaningful in CAD.10
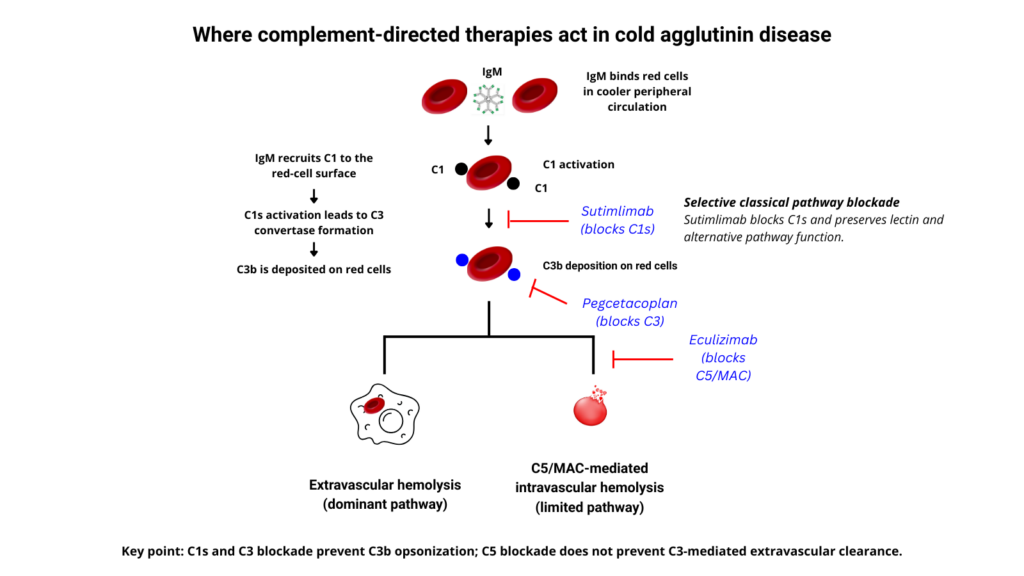
Proximal classical pathway inhibition (C1s): sutimlimab
Sutimlimab is a humanized monoclonal antibody that selectively inhibits C1s, a key upstream serine protease in the classical complement pathway.11
By blocking C1s, sutimlimab:12
- prevents activation of C4 and C3 following IgM binding
- interrupts C3-mediated red-cell opsonization
- rapidly reduces extravascular hemolysis
- preserves alternative and lectin pathway activity
Clinically, this translates into rapid suppression of complement-mediated hemolysis in many treated patients. In pivotal trials, hemolysis markers improved within days, hemoglobin rose over subsequent weeks, and mean hemoglobin increases of approximately 2–3 g/dL were observed, although normalization was not universal. Improvements in fatigue and anemia-related disease burden often paralleled hematologic response. By contrast, cold-induced circulatory symptoms were less consistently improved, consistent with the concept that these symptoms reflect IgM-mediated red-cell agglutination in cooler vascular beds rather than complement-mediated hemolysis alone.13
Importantly, these effects occur without altering antibody production or clonal persistence, reflecting intervention at the effector phase rather than the disease source.14
Why terminal complement inhibitors are not used in CAD
The success of C1s inhibition highlights an equally important negative lesson: terminal complement inhibition is biologically mismatched to CAD.
In CAD:15
- hemolysis is primarily extravascular, not intravascular
- C3 opsonization, not membrane attack complex formation, drives red-cell destruction16
- blocking C5 does not prevent C3 deposition or hepatic clearance
Terminal complement inhibitors have limited impact on the dominant chronic hemolysis of CAD because they act downstream of C3 deposition. Blocking C5 may reduce terminal pathway activation and MAC-mediated intravascular hemolysis, but it does not prevent C3b-mediated opsonization or hepatic clearance, the main pathway of red-cell destruction in CAD. It also blocks the shared terminal pathway used by classical, lectin, and alternative complement activation, creating broader terminal-pathway blockade than is needed to address the dominant CAD mechanism. Because MAC formation is an adaptive host-defense function, especially against certain encapsulated bacteria, complete terminal pathway blockade carries infectious-risk implications.17
This contrast reinforces a central principle: effective therapy in CAD must target the classical pathway proximally, at the point where disease is actually expressed.
Investigational and adjacent approaches
Investigational strategies in CAD continue to reflect the same guiding logic that underpins current therapy: interventions that block the classical pathway proximally are most likely to meaningfully alter hemolysis. Agents that act downstream, broadly, or at the terminal pathway remain mechanistically misaligned with the dominant disease biology.
Additional proximal classical pathway inhibitors are under investigation, reinforcing that the therapeutic principle is pathway targeting rather than dependence on any single agent.
What Complement-Directed Therapy Does — and Does Not — Change
Complement-directed therapy can be highly effective for complement-mediated hemolysis and anemia, but its scope is specific.
By design, proximal complement inhibition:18
- rapidly controls active hemolysis
- reduces transfusion requirements
- improves anemia and systemic symptoms
- stabilizes fluctuating or exacerbating disease
What it does not do is modify the underlying disease origin.
Complement inhibition does not:19
- suppress the pathogenic IgM clone
- reduce antibody production
- eliminate cold agglutinins
- improve cold-induced symptoms (circulatory symptoms are not mediated by complement activity)
- induce durable remission after discontinuation
When complement inhibition is stopped, hemolysis typically resumes.
This is disease control, not disease modification — a distinction that becomes critical when choosing between complement-directed and clone-directed strategies, or when sequencing them over time.
Why proximal classical pathway inhibition matters
The advantage of proximal classical pathway inhibition is timing: it interrupts the cascade before C3 deposition marks red cells for hepatic clearance.
Key advantages of proximal classical pathway inhibition include:20
- blockade of downstream C3 activation and red-cell opsonization
- preservation of alternative and lectin pathway function
- avoidance of broad or terminal complement blockade
- direct targeting of the dominant effector mechanism in CAD
This mechanism-first alignment explains why complement-directed therapy succeeds where nonspecific historical interventions often failed. Corticosteroids do not reliably interrupt classical complement activation, and splenectomy targets the wrong clearance compartment, since C3-opsonized red cells are cleared predominantly by hepatic rather than splenic macrophages.21
Speed and predictability of response
Complement inhibition produces rapid and reproducible clinical effects because it acts directly on the effector phase of hemolysis.
Across clinical studies of C1s inhibition, response characteristics are consistent with this mechanism:22
- improvement in hemolysis markers within days
- hemoglobin rise over subsequent weeks
- symptomatic improvement often preceding full laboratory normalization
- response magnitude reflects baseline complement-mediated hemolysis burden rather than clone size
This predictable tempo contrasts with clone-directed strategies, in which responses are typically delayed and depend on suppression of antibody production rather than interruption of ongoing hemolysis.23
Cold-induced circulatory symptoms are less consistently improved and should be interpreted separately from the hemolytic response. Hemolysis can improve while acrocyanosis persists. Persistent acrocyanosis or cold-induced pain does not necessarily mean complement blockade has failed; it may reflect ongoing IgM-mediated agglutination and microvascular flow impairment.
Acute exacerbations in CAD may reflect transient increases in complement availability during physiologic stress, reinforcing why proximal complement inhibition can provide rapid control of hemolysis when complement-mediated activity is dominant.24
Who benefits most
Complement-directed therapy is best suited for patients in whom active hemolysis is the dominant driver of morbidity.
Clinical contexts in which benefit is most pronounced include:25
- symptomatic anemia driven by ongoing hemolysis
- transfusion dependence
- frequent or severe exacerbations
- situations requiring rapid, predictable disease control
- contraindications to chemo-immunotherapy
This approach is particularly advantageous when disease impact is high despite a low-burden or indolent underlying B-cell clone, underscoring the dissociation between clone size and hemolytic severity in CAD.26
Case-based and expert-review literature suggest a possible role for complement inhibition in selected high-risk settings such as major surgery, but evidence is limited.
Continuous therapy and chronic disease
Because complement inhibition does not eliminate the source of pathogenic IgM, treatment is typically continuous.
Long-term use reframes CAD management from episodic intervention to chronic disease control, with several predictable implications:27
- sustained suppression of hemolysis while on therapy
- recurrence of hemolysis if treatment is interrupted
- need for long-term planning, monitoring, and shared decision-making
Extension data reinforce this principle, demonstrating maintenance of hematologic and symptomatic benefit during ongoing therapy, with deterioration toward baseline following discontinuation.28
Safety considerations
Complement-directed therapy avoids many toxicities associated with cytotoxic chemotherapy or broad immunosuppression. Its safety profile, however, reflects the biologic role of complement in host defense.
General safety principles include:29
- continued vigilance for infection
- preservation of alternative and lectin pathway activity with proximal inhibition
- a different and generally narrower infectious-risk profile than terminal complement blockade
- importance of vaccination and routine preventive care
As with all chronic therapies, safety must be weighed against disease burden, alternatives, and patient priorities.
Complement inhibition vs clone-directed therapy
Complement-directed and clone-directed therapies address different clinical questions and should not be conflated.
Complement inhibition asks:
“Can we stop hemolysis now?”
Clone-directed therapy asks:
“Can we reduce or eliminate antibody production over time?”
Because these strategies operate at different points along the disease pathway, they are not interchangeable. Inappropriate substitution of one for the other risks either delayed stabilization or unnecessary exposure to long-term therapy.
Strategic sequencing of complement-directed and clone-directed therapy is discussed in the Sequencing spoke.
Explicit limits of this strategy
Complement-directed therapy is not ideal when:30
- disease impact is minimal and observation is appropriate
- long-term continuous therapy is unacceptable to the patient
- the primary goal is disease modification rather than control
- clone-directed therapy is clearly indicated and feasible
Recognizing these limits prevents over-reliance on a powerful but incomplete therapeutic tool.
Explicit principle
Complement-directed therapy controls disease expression, not disease origin.
It is a highly effective strategy for rapidly suppressing complement-mediated hemolysis in cold agglutinin disease, but it does not eradicate the pathogenic clone or confer cure. Mastery of this strategy lies in knowing when rapid, mechanism-aligned control is the priority — and when additional or alternative approaches are required.
Reflect and Apply
A patient with CAD begins C1s inhibition. Within 10 days her hemoglobin rises and bilirubin falls, but she still develops painful acrocyanosis when exposed to cold.
Is this treatment failure?
What mechanism explains persistence of these symptoms?
What does this tell you about which disease pathway remains active?
Evidence anchor: complement-directed therapies studied in CAD
Summary derived from pivotal sutimlimab trials, extension follow-up, and limited data for terminal complement inhibition. The evidence base is strongest for C1s inhibition with sutimlimab. Evidence for C5 inhibition is more limited and mechanistically less aligned with the dominant chronic hemolytic pathway in CAD.
| Therapy / approach | Key evidence | Population / design | Main findings | Interpretation |
|---|---|---|---|---|
| Sutimlimab: CADENZA | Randomized, placebo-controlled phase 3 trial of sutimlimab in CAD.31 | 42 adults with CAD without RBC transfusion within 6 months before enrollment. Sutimlimab was given IV on day 0, day 7, then every 2 weeks for 26 weeks using weight-based dosing. | The primary composite endpoint was met in 73% of sutimlimab-treated patients versus 15% with placebo. Fatigue improved meaningfully with sutimlimab, with a greater increase in FACIT-Fatigue score than with placebo. Transfusion events and use of prohibited CAD medications were infrequent in both groups and assessed for the composite endpoint over weeks 5–26. | Demonstrates that C1s inhibition improves hemolysis, hemoglobin, and fatigue in CAD patients without recent transfusion requirement. Cold-induced circulatory symptoms were less consistently improved than hemolytic endpoints. |
| Sutimlimab: CARDINAL Part A | Open-label, single-arm phase 3 trial of sutimlimab in transfusion-dependent CAD.32 | 24 adults with CAD and at least 1 RBC transfusion within the prior 6 months. Sutimlimab was given IV on day 0, day 7, then every 2 weeks for 26 weeks using weight-based dosing. | The primary composite endpoint was met in 54% of patients. 71% were transfusion-free from weeks 5–26. Fatigue improved rapidly and meaningfully during treatment. | Demonstrates rapid and clinically meaningful control of complement-mediated hemolysis in transfusion-dependent CAD. The single-arm design limits comparison with other strategies. |
| Sutimlimab: CARDINAL Part B extension | Long-term extension follow-up of CARDINAL.33 | Continued sutimlimab exposure with median treatment duration of approximately 144 weeks, followed by post-treatment observation. | Hematologic and symptomatic benefits were sustained during ongoing treatment. After sutimlimab cessation, classical pathway inhibition was reversed, and hemolytic markers and fatigue scores returned toward pretreatment values. One patient died from CAD exacerbation approximately 1.5 months after the last dose; a separate off-treatment death from Klebsiella pneumoniae was also reported. | Supports the concept that sutimlimab provides ongoing disease control while therapy is maintained, but does not target the underlying IgM-producing B-cell clone or cold agglutinin production. Hemolysis may recur when complement blockade is withdrawn. |
| Eculizumab: C5 inhibition | Prospective, nonrandomized phase 2 data in chronic CAD and acute cold agglutinin syndrome.34 | 12 patients with chronic CAD and 1 patient with acute cold agglutinin syndrome treated with terminal complement blockade. | LDH decreased, transfusion independence improved in some patients, and hemoglobin increased modestly. Cold-induced circulatory symptoms remained unaffected. | C5 blockade acts downstream of C3 deposition and does not prevent C3b-mediated opsonization or hepatic extravascular clearance, the dominant chronic hemolytic pathway in CAD. Its effect on hemoglobin is therefore limited compared with proximal classical pathway blockade. |
Mechanistic note: Sutimlimab selectively inhibits C1s, blocking classical pathway activation upstream of C3 deposition. This prevents C3b/iC3b opsonization of red cells and thereby suppresses the dominant pathway of hemolysis in CAD. By contrast, C5 inhibition blocks terminal complement and MAC formation but does not prevent upstream C3b-mediated extravascular clearance.35
Interpretive note: The strongest clinical evidence for complement inhibition in CAD supports proximal classical pathway blockade with sutimlimab. Complement-directed therapy can rapidly control complement-mediated hemolysis, but it does not reduce pathogenic IgM production, eliminate cold agglutinins, or reliably resolve cold-induced agglutination symptoms. Modern sutimlimab-era positioning is based on drug-specific trials and contemporary expert interpretation rather than older formal CAD guideline recommendations.
Guideline perspective: How complement inhibition fits into CAD care
Guideline organizations and expert sources referenced
- British Society for Haematology (BSH)
- European expert consensus statements and reviews
- Hematology (ASH Education Program)
- British Journal of Haematology management reviews
- Regulatory guidance accompanying FDA and EMA approval of sutimlimab
Legacy guidelines (for example, BSH AIHA guideline) predate sutimlimab and primarily recommend rituximab ± bendamustine as first‑line clone‑directed therapy; more recent consensus reviews, regulatory documents, and payer/NCCN guidance now incorporate complement inhibition for symptomatic hemolytic CAD.
Shared guidance themes
- Complement-directed therapy is recommended when rapid control of hemolysis is required
- Therapy is positioned as disease control, not disease modification
- Proximal classical pathway inhibition (C1) is the mechanistically appropriate target in CAD
- Treatment selection should be guided by clinical impact and trajectory, not antibody titers or clone size
Clinical situations where complement-directed therapy is favored
Guidelines and expert reviews consistently emphasize use in patients with:
- symptomatic anemia driven by active hemolysis
- transfusion dependence
- frequent or severe exacerbations
- need for rapid, predictable disease control
- contraindications to chemo-immunotherapy
- perioperative or physiologic stress where hemolysis risk is high
Longitudinal strategy emphasized by guidelines
- Complement inhibition is typically continuous therapy
- Discontinuation is expected to result in return of hemolysis
- Long-term planning should include:
- monitoring disease control rather than cure
- anticipation of relapse if therapy is interrupted
- reassessment of goals over time
- Transition or sequencing with clone-directed therapy may be considered once stability is achieved, but is not mandatory
What guidelines emphasize
- Matching therapy to urgency and disease expression
- Mechanism-first decision-making
- Shared decision-making around long-term therapy
- Avoidance of reflexive escalation when disease is stable on treatment
What guidelines do not mandate
- Eradication of the B-cell clone
- Finite duration of therapy
- Normalization of all laboratory markers
- Use of complement inhibition in mild or minimally symptomatic disease
- A single universal strategy for all patients with CAD
Reflect & Apply
A short, judgment-focused quiz on complement-directed therapy in cold agglutinin disease.