Postscript
Introduction
- Alpha-thalassemia is an autosomal recessive inherited disorder of red blood cells. It can rarely occur as an acquired defect in disorders associated with ineffective erythropoiesis, especially myelodysplastic syndrome (MDS).
- Acquired α-thalassemia myelodysplastic syndrome (ATMDS) is HbH disease (α-thalassemia, AT) in association with a chronic myeloid disorder (usually MDS).
- ATMDS is caused by reduced synthesis of α-globin chains related to a dramatic downregulation of α-globin gene expression by inactivating mutations of the ATRX gene, which encodes the chromatin remodeling factor ATRX.
- Diagnosis is suspected in a patient with MDS and microcytosis.
- Diagnosis is confirmed by demonstrating excess α-chains form homotetramers (HbH, β4).
Definitions
- Myelodysplastic syndromes (MDS):
- A heterogeneous group of clonal hematopoietic stem cell disorders characterized by:
- Ineffective hematopoiesis and dysplasia resulting in bone marrow failure.
- Peripheral blood cytopenias.
- Increased risk of progression to acute myelogenous leukemia (AML)
- A heterogeneous group of clonal hematopoietic stem cell disorders characterized by:
- Alpha-thalassemia:
- Hereditary:
- A common inherited form of anemia that typically results from deletion of one or more of the duplicated α-globin genes on chromosome 16.
- Genotypes/phenotypes include:
- Single-gene deletion:
- α-thalassemia silent carrier status
- Asymptomatic
- Normal hematologic findings
- Two-gene deletion:
- α-thalassemia trait (minor)
- Microcytosis
- Usually no anemia
- Three-gene deletion:
- Hemoglobin H (HbH), which has four beta chains
- A type of α-thalassemia intermedia
- May cause:
- Moderate anemia
- Hepatosplenomegaly
- Jaundice
- Four-gene deletion:
- Hemoglobin Bart’s (Hb Bart’s), which has four γ chains (γ4)
- Also called Hb Bart syndrome or α-thalassemia major
- Characterized by hydrops fetalis
- Single-gene deletion:
- Hereditary:
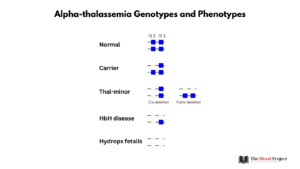
Patients with only one alpha-globin deletion (silent carrier) are asymptomatic and typically do not have anemia or microcytosis. Patients with two alpha-globin deletions (alpha-thalassemia minor or trait) in cis or trans are asymptomatic but typically have a mild microcytic anemia. Patients with three alpha-globin deletions (HbH disease) have symptomatic microcytic anemia with moderate extravascular hemolysis. Deletion of all four alpha-globin chains is not compatible with life and results in hydrops fetalis.
-
-
- Acquired (acquired HbH disease):
- Although α-thalassemia is almost always inherited, it may rarely occur as an acquired abnormality in patients with hematologic malignancy.
- Acquired (acquired HbH disease):
-
- Alpha thalassemia and mental retardation X-linked (ATRX):
- ATRX:
- An X-linked gene.
- Encodes a widely expressed protein that:
- Localizes to nuclear sub compartments called PML bodies and to pericentromeric heterochromatin, where it interacts with a known component of heterochromatin, HP1.
- Is a member of the SWI2/SNF2 family of protein
- Belongs to a family of DNA helicases/ATPases
- Has been implicated in transcriptional regulation as a chromatin remodeler, and as a transcriptional co-activator, including as a trans-acting regulator of α-globin gene expression.
- Has essential roles in development.
- ATRX loss can cause α thalassemia due to the downregulation of the α-globin gene cluster:
- Congenital/germline mutation (ATR-X syndrome):
- A severe, nonprogressive type of X-linked mental retardation.
- Rare condition that, to date, has been identified in approximately 100 families from many regions of the world.
- Clinical manifestations include:
- Intellectual disability
- Facial dysmorphias
- Hypotonia
- Skeletal anomalies
- Urogenital anomalies
- Hematological anomalies, including:
- Mild α-thalassemia
- Anemia
- Presence of HBH inclusions (HbH typically < 10%)
- Congenital/germline mutation (ATR-X syndrome):
- ATRX:
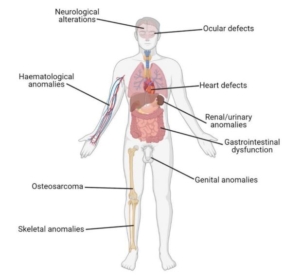
Clinical features in ATR-X syndrome. From Human Genetics (2021) 140:1625–1634.
-
-
- Acquired α-thalassemia myelodysplastic syndrome (ATMDS; aka acquired HbH disease)
- An acquired form of α-thalassemia that most commonly arises in the context of myelodysplasia
- Characterized by somatic missense mutation or splicing defect in the ATRX gene in patients with myeloid disorders, primarily MDS.
- Acquired α-thalassemia myelodysplastic syndrome (ATMDS; aka acquired HbH disease)
-
History
- White et al, 1960:
- Described 3 patients with adult acute erythroleukemia and acquired HbH.
- In their Introduction, the authors stated: “The object of the present communication is to report that under the same conditions the formation of inclusion bodies, associated with unstable hemoglobin, occurs in certain adult cases of leukemia, and to compare this phenomenon with the behavior of Hb-H”.
- The authors concluded: “There is no positive evidence for determining whether it has a genetic or acquired origin, though the latter appears possible. “
- Anagnou et al, 1983:
- Demonstrated that acquired α-thalassemia in the setting of preleukemia syndrome was characterized by decreased expression of all four α-globin genes.
- The authors posited that acquisition of a mutation that affects RNA or proteins is likely to involve in α-globin gene regulation (e.g., loss of function of a normal positive regulatory factor).
- Higgs et al, 1983:
- Reported 5 patients with hematological malignancy and acquired HbH disease (the authors refer to 34 patients previously studied by their group or described in the literature).
- In three of their 5 patients, α-chain synthesis was <10% of normal (far less than seen in patients with hereditary α-thalassemia).
- Extensive mapping with many restriction enzymes and three probes for different areas of the α-globin gene complex revealed a normal pattern with no deletions or rearrangements.
- The authors concluded: “The nature of the specific defect that results in the reduction or absence of m-chain production remains completely unknown. If it involves a major rearrangement of the DNA on chromosome 16, it does so at some distance from the &gene complex since restriction enzyme mapping shows this region to be grossly intact.”
- Gibbons et al, 1995:
- Demonstrated that ATR-X syndrome (the severe, nonprogressive type of X-linked mental retardation syndrome) results from diverse mutations of ATRX.
- Gibbons et al, 2003:
- Studied a patients with myelodysplasia associated with α-thalassemia.
- Demonstrated that:
- Reduction in α-globin expression leads to an excess of β-globin chains.
- The α/β mRNA and globin chain synthesis ratios are greatly reduced, and in the most severely
affected individuals, α chain synthesis is almost abolished, implying that all four α genes (αα/αα) are downregulated. - This degree of α-thalassemia would be lethal during development if it resulted from an inherited mutation.
- No structural abnormalities in cis to the α-globin genes have been detected.
- The downregulation of α-globin is probably associated with a trans-acting mutation.
- Used microarray analysis of highly purified (>95%) granulocytes from the peripheral blood of a newly diagnosed individual with ATMDS to search for genes whose expression might be perturbed in ATMDS.
- Using this approach, they identified a role for somatic mutations of the gene encoding the chromatin remodeling factor ATRX.
- Steensma et al, 2004:
- series of novel point mutations in ATRX detected in archival DNA samples from marrow and/or blood of
14 patients with ATMDS by use of denaturing high-performance liquid chromatography (DHPLC) - Two of the new mutations result in changes in amino acids altered in previously described pedigrees with germ line ATRX mutations (ATR-X syndrome), but the hematologic abnormalities were much more severe in the patients with ATMDS than in the corresponding constitutional cases.
- In one ATMDS case where DNA samples from several time points were available, the proportion of ATRX-mutant subclones correlated with changes in the amount of hemoglobin H
- 9 novel ATRX mutations
- series of novel point mutations in ATRX detected in archival DNA samples from marrow and/or blood of
Epidemiology
- Somatic mutations in the ATRX gene occur in ~ 0.8% of patients with MDS.
- >5:1 male–female ratio
- Reasons not understood.
- One study ruled out changes in X-inactivation as a cause of the male predominance.
- Median age at diagnosis of 68 years.
- One study reported the following low frequency of ATRX mutations in MDS:
- 0% in an unselected clinical trial cohort of 80 low risk MDS.
- 0.2–0.8% in a multicenter registry of 2,980 MDS.
- 43% of MDS with unexplained microcytosis (MCV less than or equal to 80 fL) in this same registry.
Mechanisms
- α-thalassemia is caused by a reduction in the synthesis of the α-globin chains of adult hemoglobin (HbA, α2β2) relative to β-globin synthesis, leading to excess β-globin chains, which in turn causes hemolysis and impaired erythropoiesis.
- Hereditary α-thalassemia:
- Cis-acting mutations:
- In the α-gene:
- Deletions of the α-globin genes are the most common causes.
- Point mutations are less frequent; they may be found in the:
- Structural gene
- Local cis-acting sequences, for example:
- Initiation codons
- Splice sites
- Translational termination codons
- PolyA addition sites
- In upstream regulatory elements (HS-40)
- In the α-gene:
- Trans-acting mutations:
- Inherited mutations in the ATRX gene, resulting in downregulation of α-globin gene expression (ATR-X syndrome).
- Cis-acting mutations:
- Acquired:
- Most cases result from acquired somatic mutations in the ATRX gene, causing downregulation of α-globin gene expression.
- One study reported acquired α-thalassemia and rare HbH inclusions in an MDS patient with an acquired deletion involving the telomeric region of one allele of chromosome 16, removing 2 of the 4 α-genes
- No consistent cytogenetic abnormalities have been associated with ATMDS.
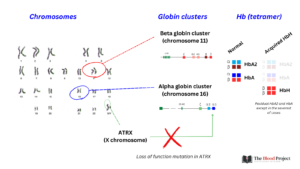
Chromosome location of alpha and beta globin clusters and the ATRX gene. Loss of function mutation of ATRX results in reduced transcription of all 4 alpha globin genes, leading to production of HbH.
Clinical presentation
- In contrast to most patients with MDS who have macrocytic or normocytic anemia, patients with α-thalassemia myelodysplastic syndrome (ATMDS) may present with severe microcytosis and hypochromia.
- In patients with acquired ATMDS, red blood cells are:
- Hypochromic
- Microcytic
- The average MCV is 75.2 fL for patients with ATMDS at the time of presentation with HbH, whereas the average MCV is 97.3 fL for MDS patients in general.
- About 25% of patients with ATMDS have MCV > 80 fL.
- Associated with striking anisopoikilocytosis
- In many cases, two distinct populations of red blood cells have been observed:
- Poorly hemoglobinized ghost cells
- Normal-appearing cells (presumably the progeny of non-thalassemic erythropoietic clones)
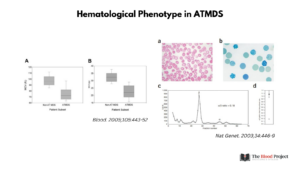
Left: Distribution of red blood cell indices in patients with ATMDS compared with a more general population with MDS. (A) MCV in ATMDS and MDS patients. Values are from the time of initial presentation with HbH or (if presentation data were unavailable) as close as possible to that time. Boxes enclose the 25th to 75th percentiles, and the middle bar denotes the median; vertical bars outline the 10th (bottom) to 90th (top) percentiles. (B) MCH in ATMDS and MDS. Boxes denote the same statistical distribution as in panel A. From Blood. 2005;105:443-452. Right: Hematological analysis in individuals with ATMDS. (a) Blood film from individual 1 showing hypochromia, anisocytosis, poikilocytosis and target cells. (b) Blood film of blood from individual 1 stained with brilliant cresyl blue showing the presence of cells with hemoglobin H (β4) inclusions. (c) Globin chain biosynthesis plot showing marked reduction in the ratio of α/β globin synthesis in individual 2. (d) A comparison of the α/β globin chain biosynthesis ratios for individual 2 (open circle) and the mean for normal controls (filled square). From Nat Genet . 2003 Aug;34(4):446-9.
Diagnosis
- Diagnostic criteria:
- Excess α-chains form homotetramers (HbH, β4). HbH demonstrated by one or more of:
- Gel electrophoresis
- High-performance liquid chromatography
- Supravital staining with brilliant cresyl blue demonstrating the presence of characteristic erythrocyte inclusions (the most sensitive technique ).
- Some form of hematologic neoplasia must be present.
- Exclude any congenital forms of thalassemia predating the hematologic neoplasm.
- Excess α-chains form homotetramers (HbH, β4). HbH demonstrated by one or more of:
- The level of HbH broadly reflects the degree of reduction in α-globin synthesis.
- Rarely, trace amounts of hemoglobin Bart’s (γ4) have been detected.
Associated hematological neoplasms
- Most commonly develops in patients with MDS.
- Other conditions include:
- di Guglielmo syndrome
- Atypical erythroid hyperplasia
- Erythroleukemia
- ALL
- de novo AML
- Myelofibrosis
- Essential thrombocythemia
Prognosis
-
- The proportion of HbH-containing cells in the peripheral blood of patients with ATMDS varies between patients, but the level in a given patient usually remains stable unless leukemia develops.
- In some patients with ATMDS, the proportion of HbH waxes and wanes during the course of the illness and sometimes disappears entirely, especially during transformation to overt acute leukemia. This observation presumably reflects somatic mosaicism and clonal evolution in MDS.
- About 25% of patients with AT-MDS will progress to AML; this risk of progression appears to be similar to that of the general MDS patient population.