Labs
Let’s begin with the patient’s complete blood count (CBC):
| WBC | Hb | Hct | MCV | MCHC | RDW-SD | PLT |
|---|---|---|---|---|---|---|
| 19.0 | 8.1 | 23.4 | 82 | 34.6 | 48.1 | 11 |
What’s what: WBC, white blood cell count; Hb, hemoglobin; MCV, mean cell volume; MCHC, mean cellular hemoglobin concentration; RDW-SD, red cell distribution width-standard deviation; platelets, PLT; Normal values: WBC 5-10 x 109/L, RBC 4-6 x 1012/L, Hb 12-16 g/dL, Hct 35-47%, MCV 80-100 fL, MCHC 32-36 g/dL, RDW-SD < 45%, platelets (PLT) 150-450 x 109/L
The patient has leukocytosis, anemia and thrombocytopenia.
A white cell differential was unremarkable, revealing an isolated neutrophilia (not shown) without evidence of a left shift. This finding is non-specific, possibly secondary to stress or bleeding.
In terms of making a diagnosis, our best bet is to focus on the anemia and thrombocytopenia. Let’s begin with a differential diagnosis for anemia (though we could just as easily begin with causes of thrombocytopenia) and then see how the concomitant thrombocytopenia helps to narrow the differential.
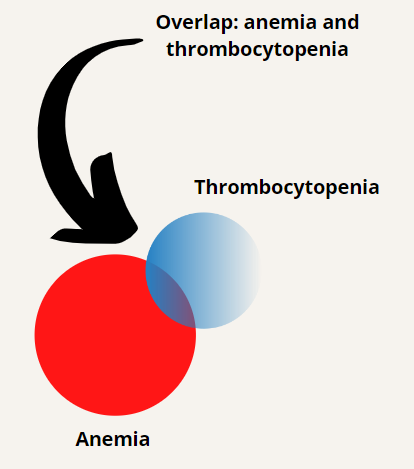
We will begin at the top of the algorithmic tree by dividing anemia into those causes associated with an appropriate reticulocyte count (we will come back to what that means in a bit) and those associated with an inappropriately low reticulocyte response. Let’s consider the side of the ledger with an appropriate reticulocyte response.
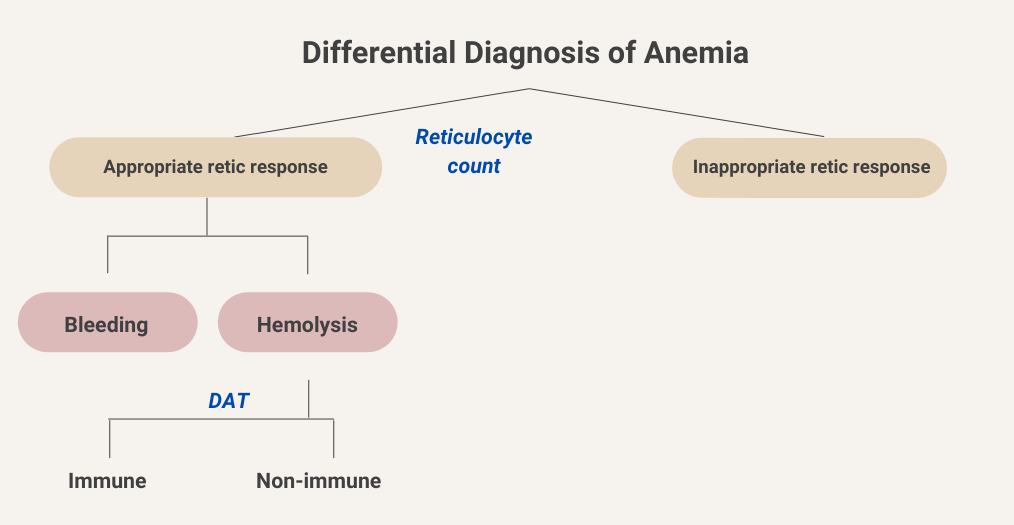
Active bleeding as a cause of anemia is usually clinically apparent from the history and/or a change in the vital signs. Hemolytic anemia can be further divided into causes that are positive for direct antiglobulin test (DAT, otherwise known as the Coombs test) and those that are DAT negative. Let’s consider DAT-positive (DAT+) vs. DAT-negative (DAT-) “buckets”.
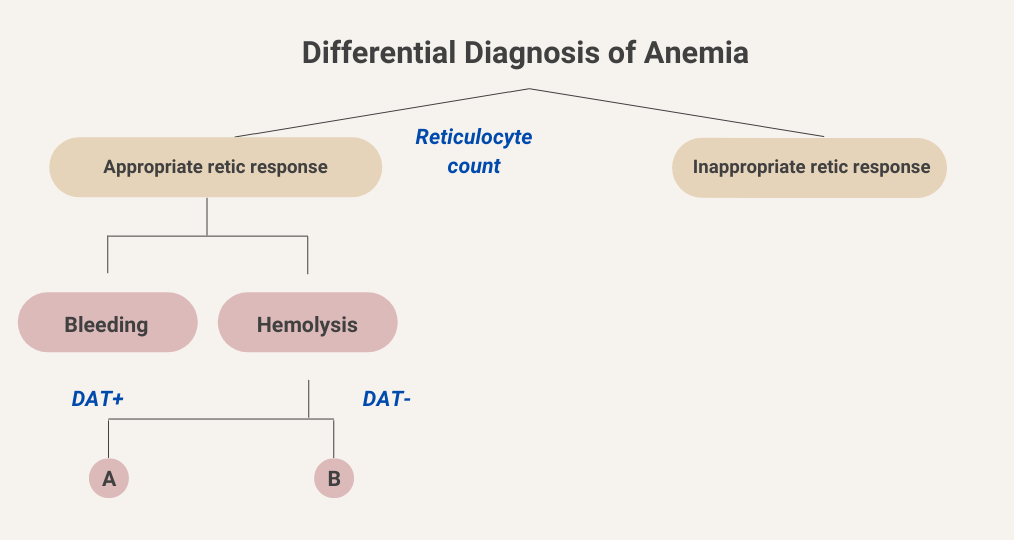
Let’s stick with the hemolysis branch for now.
In the following sorting exercise, drag the card on the top with a hemolytic disease/condition into the appropriate diagnostic bucket on the bottom.

You’re doing great! In this schematic, we have filled in the various causes of immune and non-immune hemolytic anemia. Immune-mediated (DAT-positive) hemolytic anemia may be divided into allo- and autoimmune causes. Non-immune causes are further separated into extracorpuscular and intracorpuscular etiologies. In extracorpuscular non-immune hemolytic anemia, the red bloods cell are intrinsically normal, but suffer collateral damage from their interactions with the environment, for example by passing over fibrin strands in microvessels (as occurs in thrombotic microangiopathy), by undergoing turbulent flow through paravalvular leaks (causing valve hemolysis), or by being exposed to toxins related to chronic liver disease or to certain pathogens. Intracorpuscular causes include those conditions in which the primary defect is in the red cell, in either its hemoglobin molecule, its membrane structure or its intracellular enzymes. Intracorpuscular non-immune hemolytic anemia is almost always hereditary in nature. One exception is paroxysmal nocturnal hemoglobinuria (PNH), which is an acquired red cell membrane disorder.
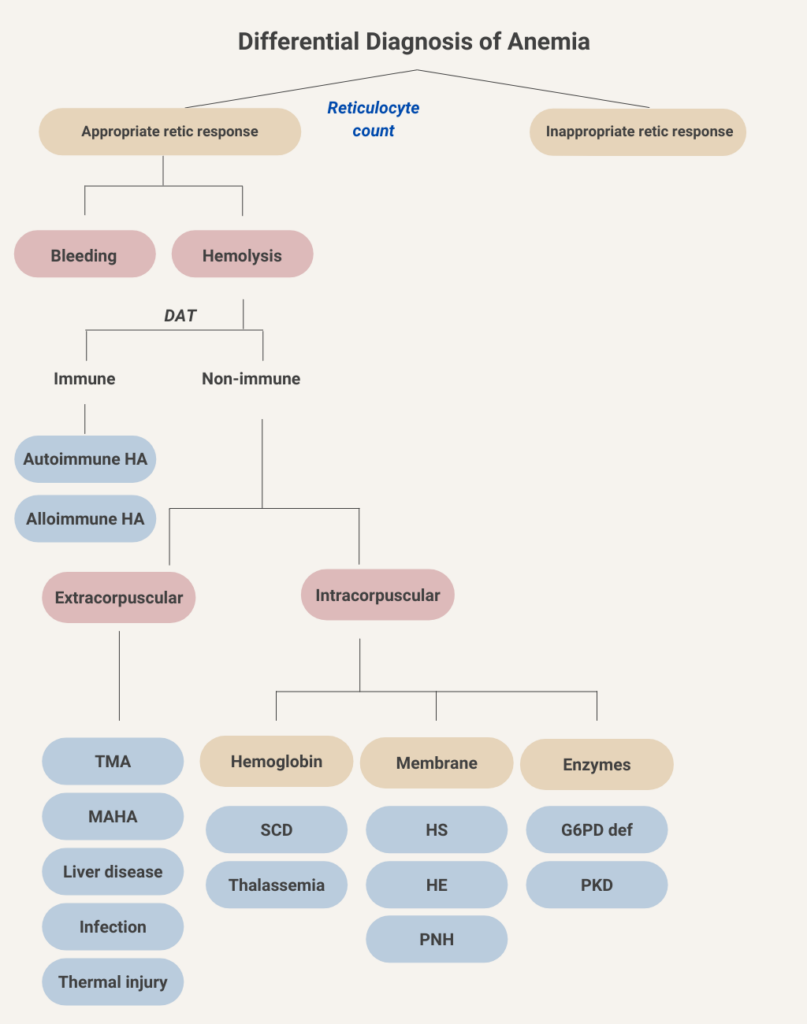
Let’s return to the first branch point in the flow chart for anemia and consider the causes of anemia associated with an inappropriate reticulocyte response (the so-called hypoproliferative anemias; upper right in above schematic).
What lab result is needed for the initial classification of hypoproliferative anemias?
Click for AnswerThe first branch point in considering hypoproliferative anemias is the mean cell volume (MCV). This represents the time-honored morphological classification of anemia and is very helpful when sorting through the differential diagnosis of anemia.
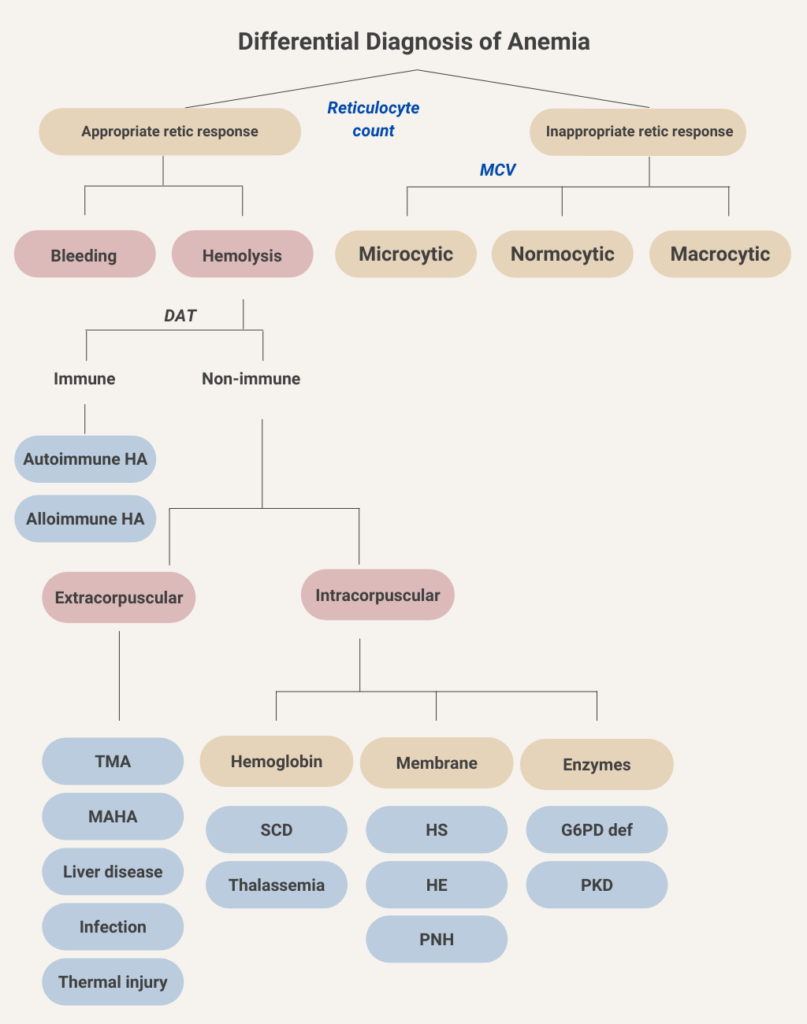
Let’s do another sorting exercise. In this case, drag the disease or condition into the appropriate red cell size bucket (the distinctions are not black and white – for example, hypothyroidism is classically associated with macrocytosis [which is the correct choice here], but in fact most such patients are normocytic).
So now the classification of anemia, based on kinetics (reticulocyte count) and morphology (mean cell volume). A second component of the morphological classification, which we have chosen not to include in order to keep things simple, is the mean corpuscular hemoglobin concentration (MCHC), which may be used to separate microcytic anemia into hypochromic (low MCHC) causes, as seen in iron deficiency anemia, and normochromic (normal MCHC) causes, which is more typical of thalassemia.
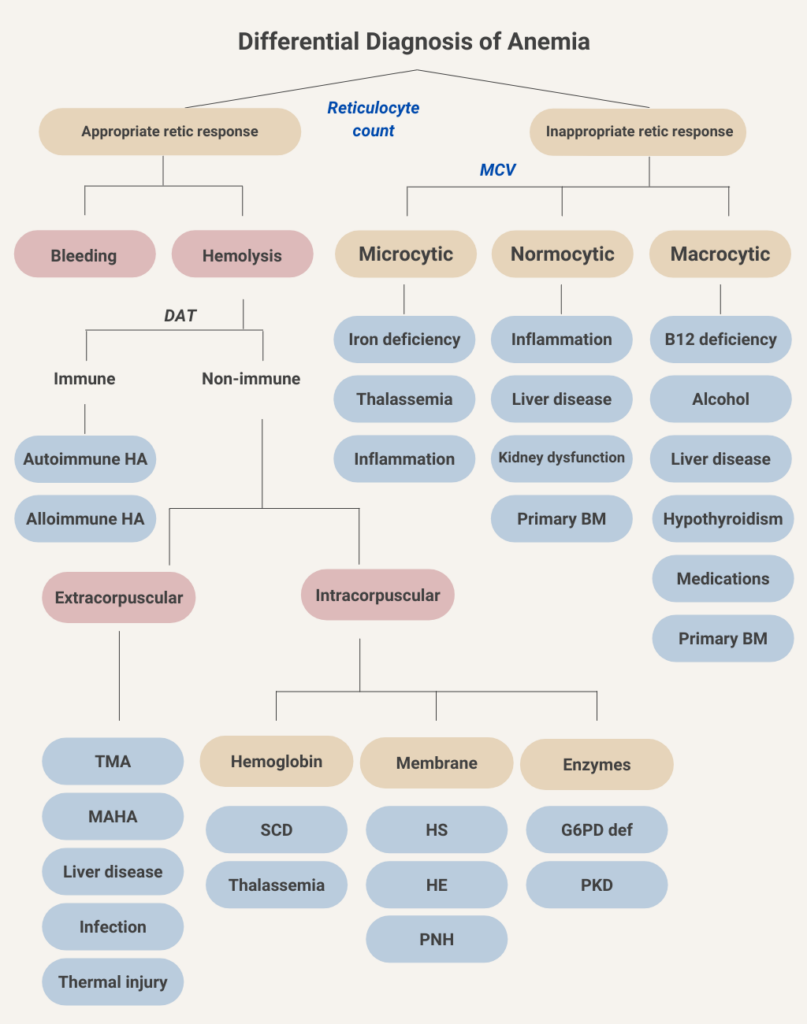
Time for another sorting exercise! This time, we are going to identify types of anemia that are typically associated with concomitant thrombocytopenia. Drag and drop the cause of anemia on the top cards into the appropriate bucket (associated with concomitant thrombocytopenia or not).
The following is the differential diagnosis for anemia, with those conditions associated with concomitant thrombocytopenia indicated in dark blue. We have included patients with bleeding because thrombocytopenia may cause of bleeding, and because massive transfusion may lead to dilutional thrombocytopenia. Although rare, patients with severe iron deficiency anemia may have low platelet counts (not shown), and those with autoimmune hemolytic anemia may have concomitant immune thrombocytopenia (Evans syndrome, also now shown).
You should be a pro by now: what lab test would help you separate hemolytic from non-hemolytic causes of anemia?
Click for AnswerW
Let’s look at the reticulocyte count in our patient:
| Parameter | Value |
|---|---|
| Percentage reticulocytes | 4.5% |
| Absolute reticulocyte count | 0.13 x 1012/L |
What is the patient’s red cell count in trillions (1012)/L?
Correct! 4.5% x [2.89 x 1012/L] = 0.13 x 1012/L
Let’s look at the various markers of hemolysis when the patient first presented to the emergency room:
| Parameter | Value | Normal Range |
|---|---|---|
| LDH | 2112 IU/L | 94-250 IU/L |
| AST/ALT | 71/30 IU/L | 0-40 IU/L for both |
| Haptoglobin | < 10 mg/dL | 30-200 mg/dL |
| Bilirubin (total) | 3.6 mg/dL | 0-1.5 mg/dL |
In summary, the above results are classic for hemolysis!
The peripheral smear from the patient is shown here:
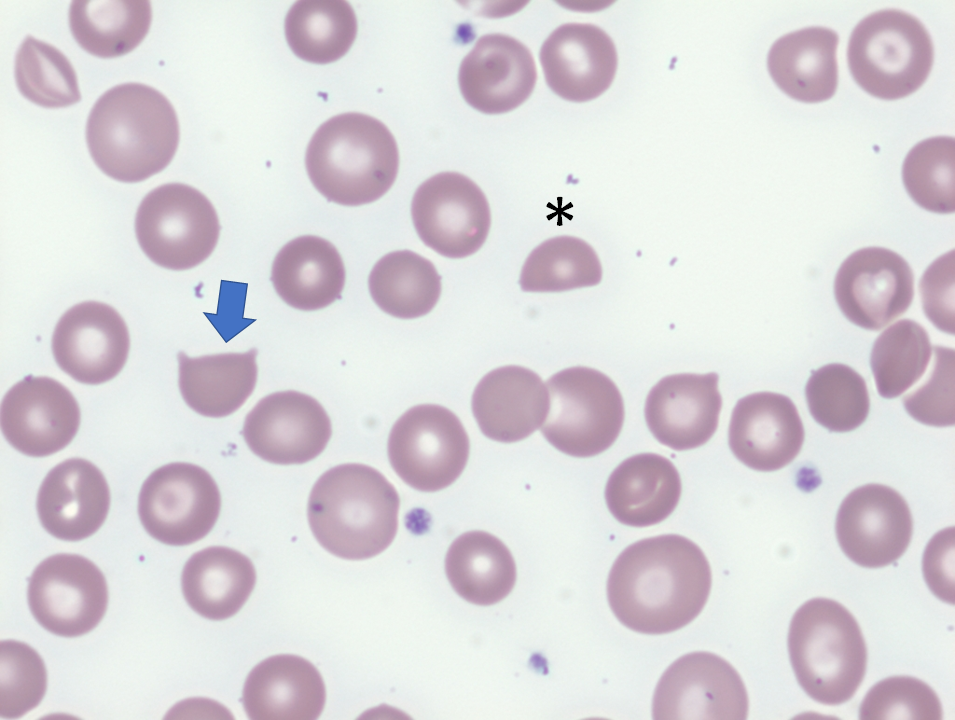
The patient’s direct antiglobulin test (DAT) was negative.
The combination of appropriately elevated reticulocytes, positive hemolytic makers and negative DAT places him in the extracorpuscular hemolytic anemia “bucket”. The presence of schistocytes on the peripheral smear further narrows the diagnosis to thrombotic microangiopathy (TMA), which may be caused by thrombotic thrombocytopenia purpura, hemolytic uremic syndrome, disseminated intravascular coagulation or a handful of other secondary causes (see next slide),
Let’s consider the thrombotic microangiopathies (TMAs) for a moment.
Definition: TMAs are a group of rare disorders characterized by thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and microvascular thrombus formation (fibrin/platelet rich), leading to tissue injury. MAHA, in turn, is characterized by red blood cell destruction within the microvasculature accompanied by thrombocytopenia due to platelet activation and consumption.
Classification: TMAs may be classified as primary or secondary. Primary TMAs occur spontaneously with no associated underlying cause, and include thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Secondary TMAs are associated with an underlying condition.
PLASMIC score

Notes:
- The higher the score, the greater the likelihood of TTP.
- Many of the parameters included in the PLASMIC score are negative predictors of thrombotic thrombocytopenic purpura (TTP). For example, the score takes into consideration non-TTP thrombotic microangiopathy caused by transplantation and cancer (based on history), disseminated intravascular coagulation (based on an elevated INR) and hemolytic uremic syndrome (based on elevated creatinine).
- Schistocytes are not included in the score because they are considered a precondition for applying the score.
- The PLASMIC score was developed using a cohort of 214 patients with suspected thrombotic microangiopathy and has been validated in 2 additional cohorts of patients.
Our patient’s platelet count of 11 x 109/L qualifies for one point. He also gets one point for the unmistakable finding of hemolysis, and another point each for not having a history of cancer or solid organ or hematopoietic stem cell transplantation. That’s four points so far. Add another point for the normal PT/INR and we are up to a total of five points. The patient’s mean cell volume (MCV) was < 90 fL for another point. Finally, the creatinine was normal, bringing the tally to seven points for a pretest probability of about 70%.
The patient is admitted directly to the hospital
