About the Condition
Description/definition:
Hemophagocytic lymphohistiocytosis (HLH) is a multisystem, life-threatening clinical syndrome of unchecked immune activity, increased secretion of cytokines, and persistent activation of cytotoxic T lymphocytes and natural killer (NK) cells leading to macrophage activation, systemic inflammation, and organ damage.
HLH is traditionally classified as:
- Primary or familial: driven by genetic mutations in proteins required for normal T-cell / NK cell function
- Secondary or acquired: caused by infection, malignancy, or autoimmune disease

Pathophysiology:
The pathogenesis of HLH arises from an inability of the immune system to adequately restrict or terminate stimulation of the system by various possible triggers. In primary HLH, this dysfunction is driven by mutations that impact the function of granule-mediated cytotoxicity. This immune dysfunction is acquired in secondary HLH in response to underlying autoimmunity, infection, or malignancy. Unbridled antigen presentation leads to persistent immune response with the activation of interferon-g producing T cells. The subsequent result is hypersecretion of proinflammatory cytokines and high levels of macrophage activation leading to hemophagocytosis.
Infections are the most common trigger of HLH. Among infectious etiologies, EBV is the most common trigger. Other viral causes include CMV, HIV, herpes viruses, hepatitis viruses, influenza, and parvovirus B19. Of bacterial infections causing HLH, tuberculosis is the most common. Parasites and fungi are less commonly reported – among them, histoplasma, plasmodium, leishmania, and toxoplasma are the most frequently identified.
Malignancies, primarily lymphomas, are also a significant cause of HLH. These lymphomas have been shown to produce inflammatory cytokines as the initial stimulus for T cell activation. Solid tumors are a rare but reported underlying condition in HLH.
In patients with autoimmune disease, acquired abnormalities in immune function are the suspected pathophysiology of HLH development.
Diagnosis:
There is no clinical, laboratory, or histopathologic finding alone that is pathognomonic for the diagnosis of HLH. Diagnosis is guided by clinical scoring systems including the:
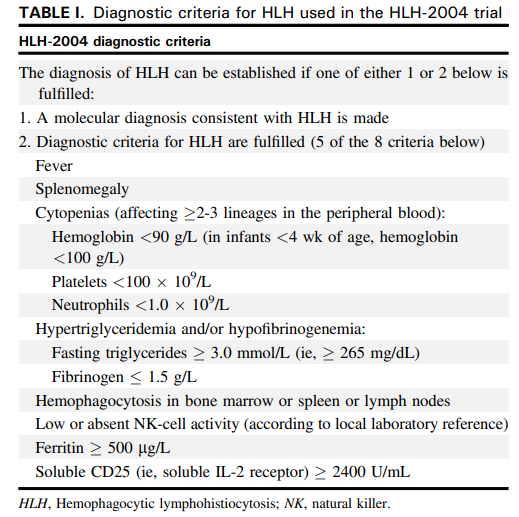
Or molecular diagnosis based on pathologic mutations in PRF1, UNC13D, STXBP1, RAB27A, STX11, SH2D1A, or XIAP.
In addition to the lab work listed above and bone marrow biopsy, patients should undergo broad infectious work-up, investigation of autoimmune etiologies as suggested by history and physical exam, and further imaging/biopsy if malignancy is suspected.
Treatment:
Treatment guidelines for HLH are primarily derived from the pediatric population as there are no prospective trials for first-line treatment for HLH in adults. The goal of therapy in HLH is to terminate the uncontrolled immune response with immunosuppressive and myelosuppressive agents. This is primarily achieved with high-dose corticosteroids and etoposide as defined by the HLH-94 trial. In patients with neurologic symptoms, intrathecal methotrexate is added to the regimen.
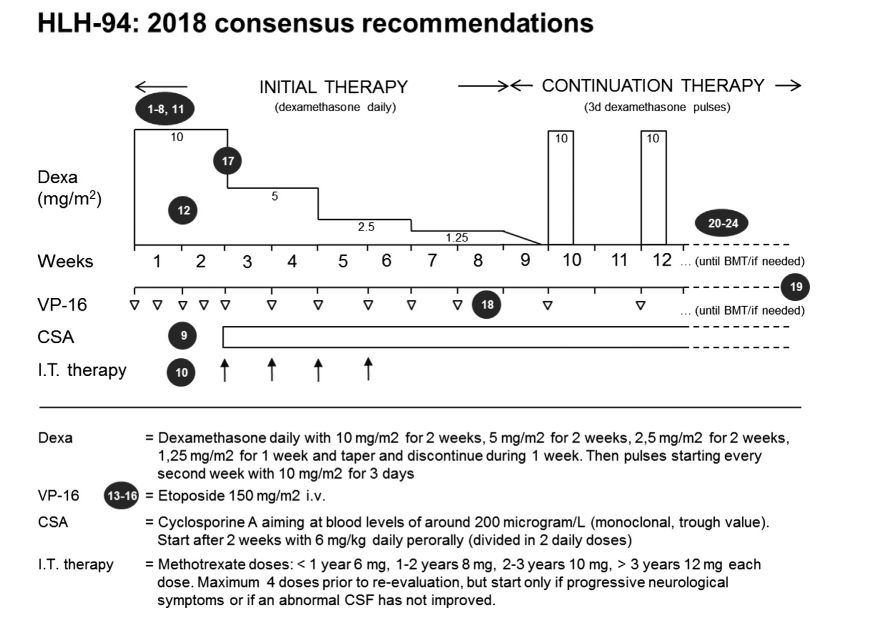
Patients with familial/primary HLH or those that relapse after initial therapy ultimately proceed to allogeneic stem cell transplant.
In addition to these immunosuppressive therapies, treatment should be addressed at the underlying cause (i.e., antimicrobial therapy for infection, disease-specific therapy for malignancy/autoimmunity).
There are ongoing studies to further refine HLH therapy. Among them are the HLH-2004 trial which modifies the HLH-94 regimen by adding cyclosporine.
Salvage therapy:
There is data to support a combination of doxorubicin, etoposide, and methylprednisolone for refractory adult HLH patients. Another modified regimen includes the addition of PEG-asparaginase. The agent emapalumab, a monoclonal antibody directed at interferon-γ is under active investigation in the treatment of pediatric patients with refractory HLH.