What if anemia of inflammation is not a cost, but a circulatory adjustment?
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
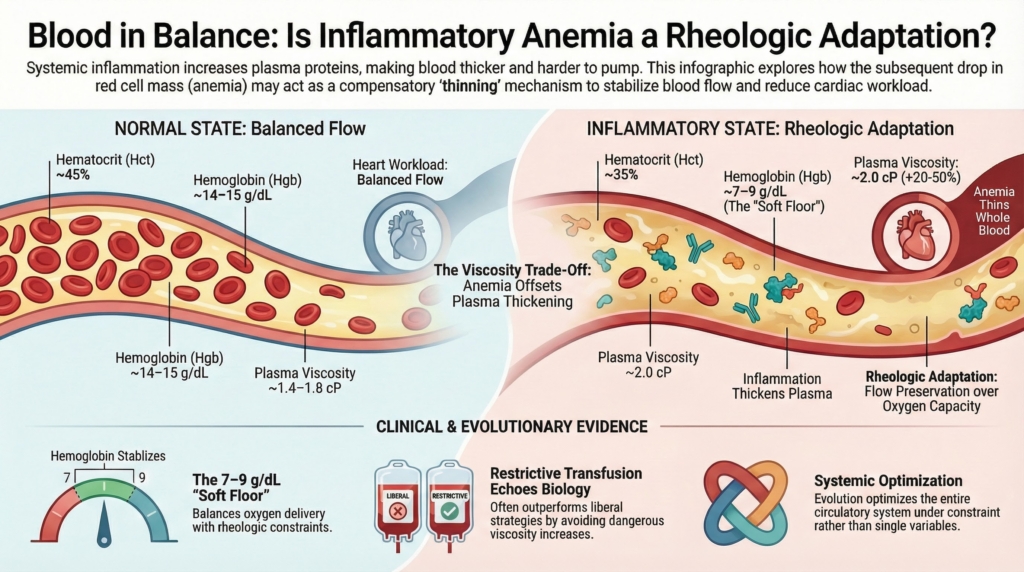
Inflammation alters the rheology of blood.
Fibrinogen rises. Immunoglobulins accumulate. Acute-phase proteins crowd the intravascular space. Plasma becomes measurably more viscous.
Viscosity governs flow.
At the same time, hemoglobin falls, and with it, hematocrit.
Clinicians think in hemoglobin because oxygen delivery is the obvious concern.
But blood flow responds primarily to hematocrit, the fraction of blood volume occupied by red cells.
Oxygen content determines carrying capacity.
Viscosity determines resistance.
Inflammation increases one.
Anemia decreases the other.
These changes may not be independent. They may together constrain whole-blood viscosity within a narrower band than either would alone.
The evolutionary frame
In ancestral environments, systemic inflammation meant infection or tissue injury. The host response required rapid protein synthesis, clot containment, immune activation, and iron restriction. All of these increase plasma protein burden.
But increasing plasma protein concentration thickens blood. Increased viscosity raises vascular resistance, increases cardiac workload, destabilizes microvascular flow, and may contribute to thrombotic risk in inflammatory states.
If hematocrit remained fixed at pre-inflammatory levels, circulation would operate in a mechanically stressed state.
Lowering hematocrit thins blood.
The conventional interpretation is that anemia is a cost paid for iron sequestration. But evolution does not optimize single variables. It optimizes systems under constraint.
If a reduction in red cell mass partially offsets protein-driven plasma thickening while preserving adequate oxygen transport, the net effect may be stabilizing rather than sacrificial.
That is a quantitative claim.
Quantitative check: are the magnitudes commensurate?
Normal plasma viscosity: ~1.4–1.8 cP
Typical inflammatory increase: commonly on the order of ~20–50%
Example: 1.5 → 2.0 cP
This is not a 10-fold or 100-fold change. It is a tens-of-percent perturbation.
In paraproteinemic states, particularly IgM excess, plasma viscosity may rise to ~4–5 cP or higher, roughly 2–3× baseline, which is where hyperviscosity symptoms appear. Ordinary acute inflammation produces smaller shifts.
Now consider hematocrit.
A drop from 45% to 35%, common in inflammatory anemia, produces a disproportionately large reduction in whole-blood viscosity because hematocrit exerts a steep nonlinear influence on flow resistance.
At moderate to high shear rates, a 10-point hematocrit reduction can offset a substantial fraction of a 30–50% rise in plasma viscosity.
The scales are comparable.
The fall in hematocrit is not trivial relative to the rise in plasma protein burden. It is of sufficient magnitude to plausibly bring whole-blood viscosity closer to baseline.
The “soft floor” phenomenon
In many critically ill patients without active bleeding, hemoglobin stabilizes in the 7–9 g/dL range, whether through biology, clinical restraint, or both.
Below that range:
- cardiac output must rise steeply
- myocardial oxygen demand increases
- tissue oxygen extraction becomes strained
The system appears to tolerate moderate anemia but resist severe anemia.
One possibility is that this floor reflects intersecting constraints: oxygen delivery limits, energetic cost, and rheologic balance. The mechanism is unknown, but the bounded range is real.
Evolution rarely produces rigid set points. It produces bounded recalibration.
Inflammatory anemia may represent a shift to a new equilibrium rather than progressive injury.
Restrictive transfusion as physiologic echo
Randomized trials in general ICU populations show that transfusing to higher hemoglobin targets often does not improve survival compared with restrictive strategies.
Transfusion increases oxygen-carrying capacity.
It also increases hematocrit and viscosity.
If inflammatory anemia helps stabilize rheology in a protein-rich environment, liberal transfusion may work against that stabilizing process in some settings, even if in others higher hemoglobin targets are beneficial.
This does not prove the mechanism.
But it aligns with it.
A rheologist might object
- Low-shear microcirculation behaves differently. Fibrinogen increases red cell aggregation and yield stress; anemia may not fully normalize low-shear rheology.
- Plasma viscosity changes are modest in many inflammatory states. The hematocrit decline may exceed what is strictly required for viscosity compensation, implying additional biological drivers.
- No known viscosity sensor exists. The molecular pathways of inflammatory anemia are well characterized, but a direct rheologic feedback mechanism has not been identified.
Even if iron sequestration and pathogen control are dominant drivers, viscosity homeostasis could still represent a secondary but physiologically relevant pressure influencing how far hematocrit falls.
These objections do not invalidate the hypothesis.
They define what must be measured.
How this could be tested
A straightforward prospective study could determine whether inflammatory anemia contributes to viscosity homeostasis.
Population
Hospitalized patients with systemic inflammation and no active bleeding.
Serial measurements
- plasma viscosity
- whole-blood viscosity at high and low shear
- fibrinogen and immunoglobulin burden
- hemoglobin and hematocrit trajectories
Key questions
- Within individual patients, does rising plasma viscosity correlate temporally with falling hematocrit?
- Is high-shear whole-blood viscosity constrained relative to what would be expected if hematocrit remained constant?
- Does transfusion measurably increase whole-blood viscosity in protein-rich inflammatory states?
A complementary ex vivo experiment could hold plasma constant and vary hematocrit, directly measuring how much hematocrit reduction offsets inflammatory plasma thickening.
These are tractable experiments.
The hypothesis is not philosophical.
It is measurable.
A reframing
Inflammation thickens plasma.
Anemia thins blood.
Seen separately, they appear antagonistic.
Seen together, they may represent coupled regulation.
Anemia of inflammation may not simply be an evolutionary trade-off tolerated for pathogen control. It may be part of a coordinated circulatory recalibration in a protein-dense inflammatory milieu.
The body may not be sacrificing oxygen delivery recklessly.
It may, in part, be preserving flow under altered rheologic constraints.