Mechanism defines the map. Clinical reality defines its limits.
Note: The video and audio linked above were generated with the assistance of AI. Clinical accuracy has been reviewed, but no AI-generated content can be guaranteed to be fully error-free.
Why this matters
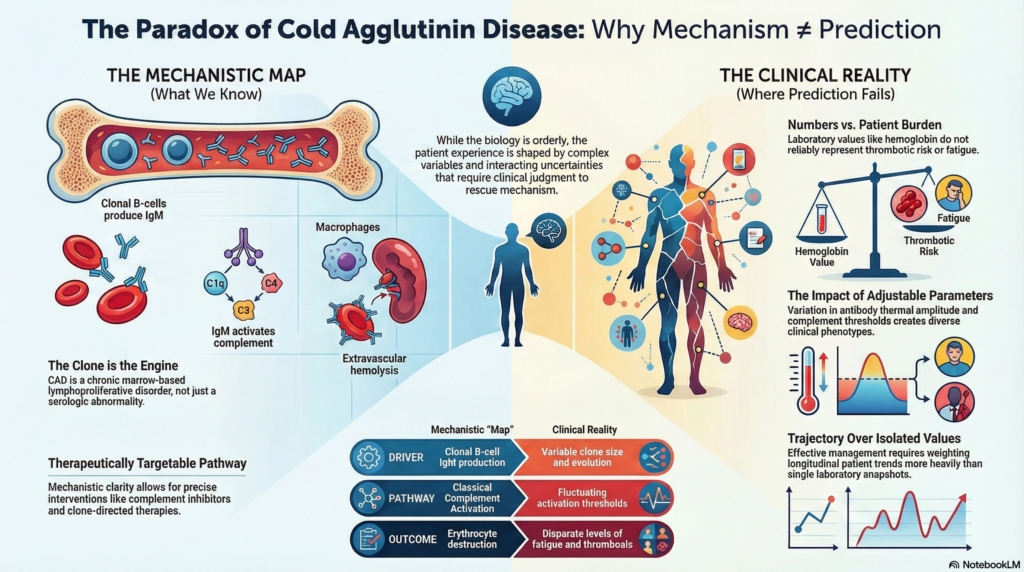
Cold agglutinin disease is one of the clearest examples in hematology of a disorder whose mechanism is well defined yet whose clinical behavior remains unpredictable.1
Its biology is orderly. Its clinical expression is not.
CAD therefore teaches a lesson that applies across medicine: understanding mechanism does not equal predicting course.
In a curriculum that emphasizes mechanistic reasoning, this is the necessary counterweight. Mechanism is indispensable, but it is not sufficient.
Earlier essays in this curriculum showed how mechanism rescues pattern when pattern misleads; this essay shows the reciprocal truth, that judgment must rescue mechanism when mechanism reaches its limits.
What we know with confidence about pathogenesis
The core biologic sequence is stable and reproducible. A clonal marrow B-cell population produces cold-reactive monoclonal IgM. At lower temperatures, the antibody binds erythrocytes, activates the classical complement pathway, and deposits complement fragments on the red-cell surface. When blood returns to warmer circulation, IgM dissociates but complement remains, marking cells for removal. Macrophages progressively strip membrane from these tagged erythrocytes until they are cleared, producing predominantly extravascular hemolysis.2
This pathway is experimentally validated, physiologically coherent, and therapeutically targetable.3
The mechanistic backbone is clear.
That clarity is precisely what makes the remaining uncertainty so instructive.
Where prediction fails despite mechanistic clarity
If mechanism alone determined disease expression, patients with CAD would behave similarly. They do not.4
Some have mild anemia and intermittent symptoms. Others develop severe hemolysis, disabling circulatory manifestations, thrombosis, or recurrent exacerbations. The steps of the pathway are the same. The intensity, timing, and modulation of those steps are not.5
Variation arises because the system contains multiple adjustable parameters:
- complement activation thresholds differ between individuals, shaped by baseline complement availability, regulatory proteins, inflammatory state, and physiologic stressors that shift the system closer to, or farther from, activation
- antibody properties vary in thermal amplitude, binding strength, and specificity
- the underlying clone differs in size, biology, and evolution
- environmental triggers intermittently amplify the pathway
- host regulatory systems modify downstream effects
The mechanism is uniform. The modifiers are not.
CAD therefore illustrates a general principle of biologic systems:
Predictability declines as interacting variables increase, even when the governing mechanism is known.
The clonal foundation
CAD is not simply an antibody phenomenon. It is a clonal lymphoproliferative disorder expressed through an autoimmune mechanism.6
That distinction matters.
Reframing CAD as a marrow-based clonal disease rather than a serologic abnormality clarifies why:
- the condition is chronic rather than transient
- responses to therapy vary in durability
- clone-directed treatments can be effective
- laboratory antibody measurements alone do not define disease behavior
The antibody is the mediator.
The clone is the engine.
Just as serology alone does not capture the disease’s biology, laboratory values alone do not capture its clinical impact.
When numbers diverge from burden
One of the most clinically important lessons of CAD is that laboratory values do not reliably represent disease impact.
Thrombotic risk is increased in CAD, yet it does not track with anemia severity. Patients with modest hemoglobin reductions may still develop serious vascular complications.7
Fatigue and cold-induced functional impairment can be profound even when hemoglobin appears stable. LDH may rise before hemoglobin changes. Circulatory symptoms may worsen despite reassuring counts.8
Laboratory magnitude is not clinical magnitude.
Numbers measure selected outputs of a system. Patients experience the system itself.
Why prognosis must be probabilistic
CAD is often chronic and compatible with long survival, yet individual trajectories vary widely. Some patients remain stable for years. Others experience episodic worsening, progressive symptom burden, or evolving disease behavior.9
This variability reflects interacting uncertainties:
- triggers are unpredictable
- complement activity fluctuates
- clonal biology evolves
- exposures differ
- responses to therapy vary
For such systems, prognosis cannot be deterministic. It must be probabilistic.
This is not a limitation of knowledge. It is a property of complex biology.
Therapeutic clarity and therapeutic limits
Mechanistic understanding has transformed treatment. Complement inhibitors can rapidly suppress hemolysis and improve symptoms, validating the central role of classical complement activation.10
Yet therapeutic success exposes therapeutic limits.
If complement blockade improves disease but relapse occurs when treatment stops, then therapy is controlling a downstream process while leaving the upstream driver intact.
Complement inhibition targets the effector pathway and can rapidly improve hemolysis and fatigue, but it is usually a control strategy rather than a cure. Clone-directed therapy targets the clonal source and can produce durable remissions, but responses are slower, not universal, and may not address acute complement-driven instability in the moment.11
Effective management therefore requires matching treatment strategy to phenotype rather than to diagnostic label alone.
Boundaries of current knowledge
Even in a mechanistically elegant disease, some uncertainties reflect gaps in knowledge, and others reflect limits of predictability in complex systems:
- Why do some patients with similar serologic profiles have vastly different clinical severity?
- What determines complement activation thresholds in vivo?
- Which clonal features predict disease behavior?
- Which patients benefit most from complement inhibition versus clone-directed therapy?
- What signals indicate impending clinical worsening?
These are not failures of science. Some reflect gaps that better data may close. Others mark boundaries where prediction remains probabilistic because the system itself is inherently variable.
Teaching uncertainty without paralysis
Uncertainty is not an obstacle to clinical reasoning. It is its working condition, and it creates predictable traps: overtreatment to relieve discomfort, undertreatment to avoid being wrong, and inertia when reassessment fails to translate into action.
In CAD, good practice depends on:
- reassessing patients longitudinally rather than assuming stability
- recognizing trigger-driven changes rather than attributing all fluctuation to “the disease”
- weighting trajectory more heavily than isolated values
- matching intervention intensity to observed behavior
The task is not to eliminate uncertainty. It is to act responsibly within it.
This requires a specific cognitive stance: decisiveness without false certainty.
Explicit principle
Do not mistake mechanistic clarity for clinical predictability.
Cold agglutinin disease is mechanistically elegant yet clinically variable. It shows that medicine advances not when uncertainty disappears, but when clinicians learn to reason skillfully at its boundary.
Key points
- mechanism is not prediction: CAD has a stable pathway, but variable expression because modifiers shift intensity and timing.
- trajectory beats snapshot: trends (LDH, symptoms, exacerbations) often carry more signal than a single hemoglobin value.
- numbers are partial outputs: symptom burden and vascular risk can diverge from anemia severity.
- judgment is the skill: act decisively without false certainty, reassess longitudinally, and match intervention intensity to observed behavior.
Reflect and Apply
A patient with confirmed CAD has stable hemoglobin, rising LDH, new acrocyanosis, and worsening fatigue.
Which signal should guide treatment escalation: laboratory trend, symptom change, or disease label? Why?
Before deciding, ask which signal reflects trajectory, which reflects mechanism, and which reflects lived physiology, then privilege the signal that best captures changing system behavior.